

A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair
- PMID: 27264673
- PMCID: PMC5134331
- DOI: 10.1002/ajmg.a.37781
A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair
Abstract
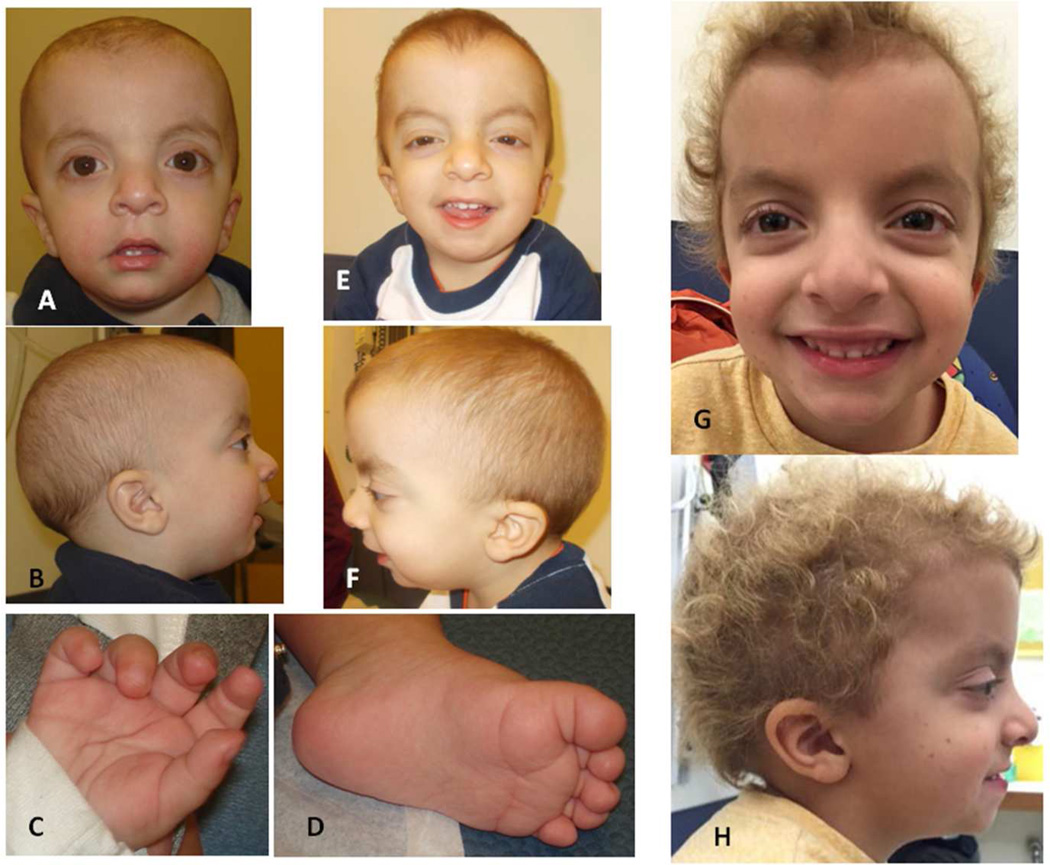
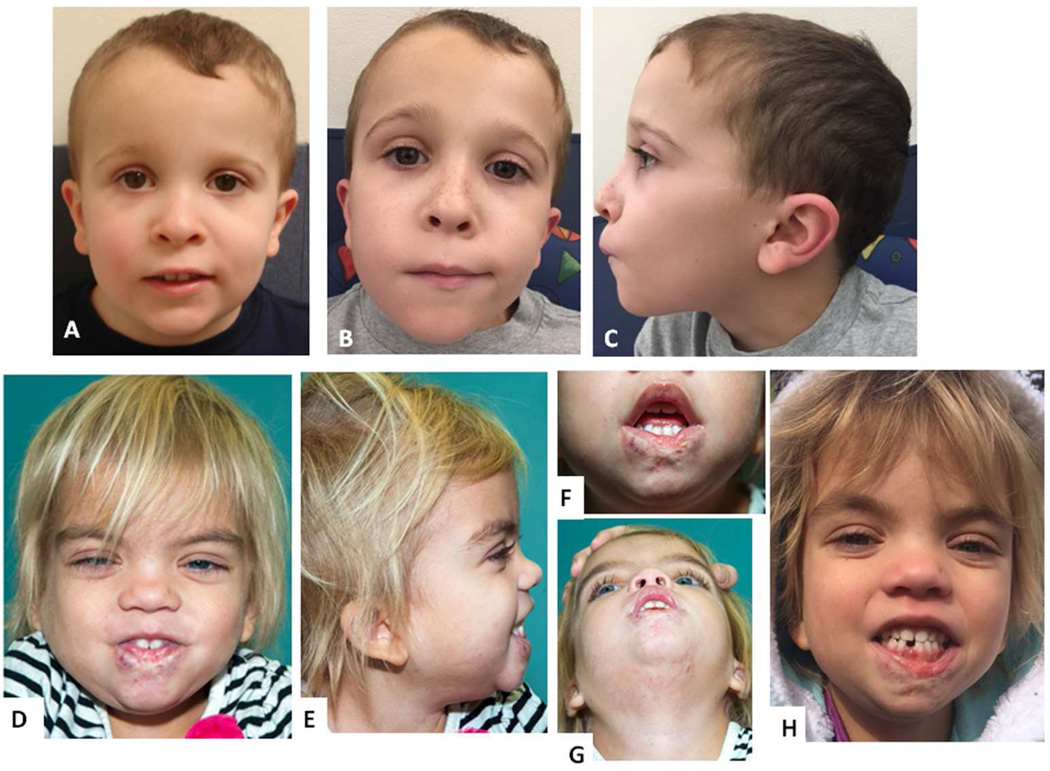
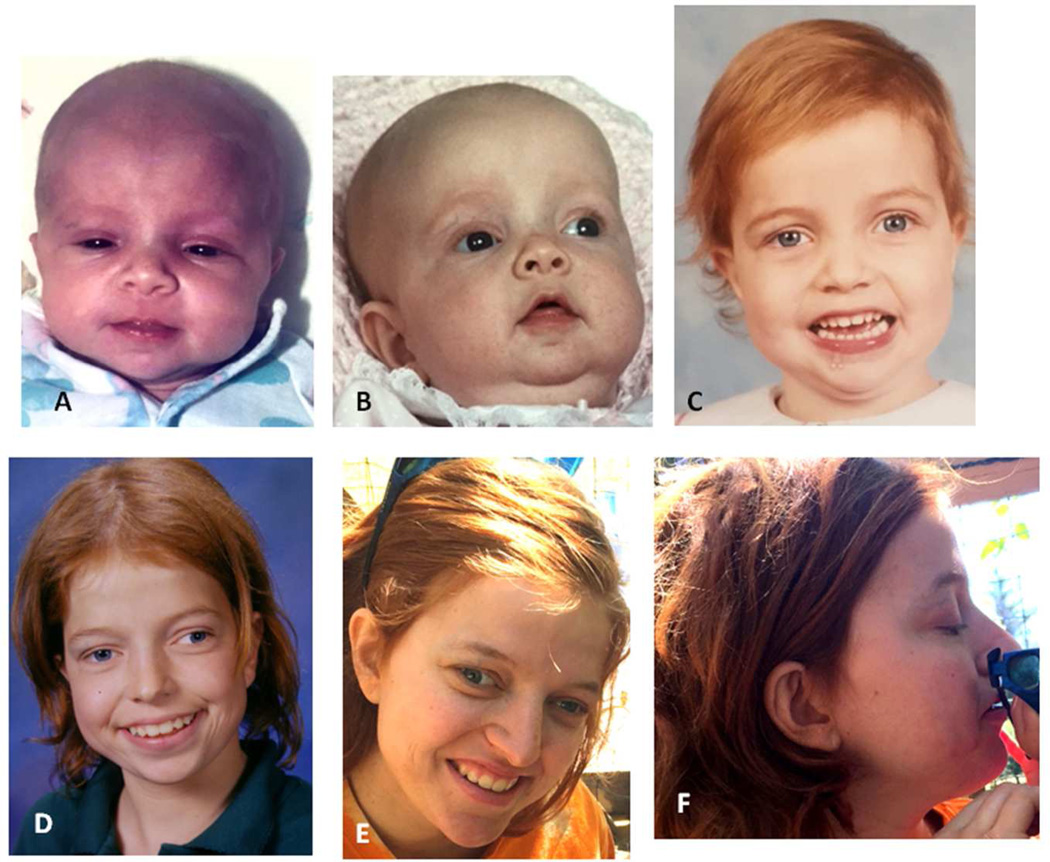
Noonan syndrome is a rasopathy caused by mutations in multiple genes encoding components of the RAS/MAPK pathway. Despite its variable phenotype, limited genotype-phenotype correlations exist. Noonan syndrome with loose anagen hair (NS-LAH) is characterized by its distinctive hair anomalies, developmental differences, and structural brain abnormalities and is caused by a single recurrent missense SHOC2 mutation. SHOC2 forms a complex with protein phosphatase 1 (PP1C). Protein phosphatases counterbalance kinases and control activation of signaling proteins, such as the mitogen-activated protein kinases of the RAS/MAPK pathway. Here we report four patients with de novo missense mutations in protein phosphatase one catalytic subunit beta (PPP1CB), sharing a recognizable phenotype. Three individuals had the recurrent PPP1CB c.146G>C, p.Pro49Arg mutation, the fourth had a c.166G>C, p.Ala56Pro change. All had relative or absolute macrocephaly, low-set and posteriorly angulated ears, and developmental delay. Slow growing and/or sparse hair and/or an unruly hair texture was present in all. Three individuals had feeding difficulties requiring feeding tubes. One of two males had cryptorchidism, another had pectus excavatum. Short stature was present in three. A female with the recurrent mutation had a Dandy-Walker malformation and optic nerve hypoplasia. Mild ventriculomegaly occurred in all, cerebellar tonsillar ectopia was seen in two and progressed to Chiari 1 malformation in one individual. Based on the combination of phenotypic findings and PPP1CB's effect on RAF dephosphorylation within the RAS/MAPK pathway, this novel condition can be considered a rasopathy, most similar to NS-LAH. Collectively, these mutations meet the standardized criteria for pathogenicity. © 2016 Wiley Periodicals, Inc.
Keywords: Dandy-Walker malformation; Noonan syndrome; PPP1CB; developmental delay; loose anagen hair; rasopathy; short stature.
© 2016 Wiley Periodicals, Inc.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Abraham D, Podar K, Pacher M, Kubicek M, Welzel N, Hemmings BA, Dilworth SM, Mischak H, Kolch W, Baccarini M. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J Biol Chem. 2000;275:22300–22304. - PubMed
-
- Baldassarre G, Mussa A, Banaudi E, Rossi C, Tartaglia M, Silengo M, Ferrero GB. Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation. Am J Med Genet A. 2014;164:3120–3125. - PubMed
-
- Beier AD, Barrett RJ, Burke K, Kole B, Soo TM. Leopard syndrome and Chiari type I malformation: A case report and review of the literature. Neurologist. 2009;15:37–39. - PubMed
-
- Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, Lipzen A, Zampino G, Mazzanti L, Diglio MC, Martinelli S, Flex E, Lepri F, Bartholdi D, Kutsche K, Ferrero GB, Anichini C, Selicorni A, Rossi C, Tenconi R, Zenker M, Merlo D, Dallapiccola B, Iyengar R, Bazzicalupo P, Gelb BD, Tartaglia M. Mutation of SHOC2 promotes aberrant protein N-myristolation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41:1022–1026. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
