

IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly
- PMID: 27264908
- PMCID: PMC4893743
- DOI: 10.1038/srep27459
IVA cloning: A single-tube universal cloning system exploiting bacterial In Vivo Assembly
Abstract
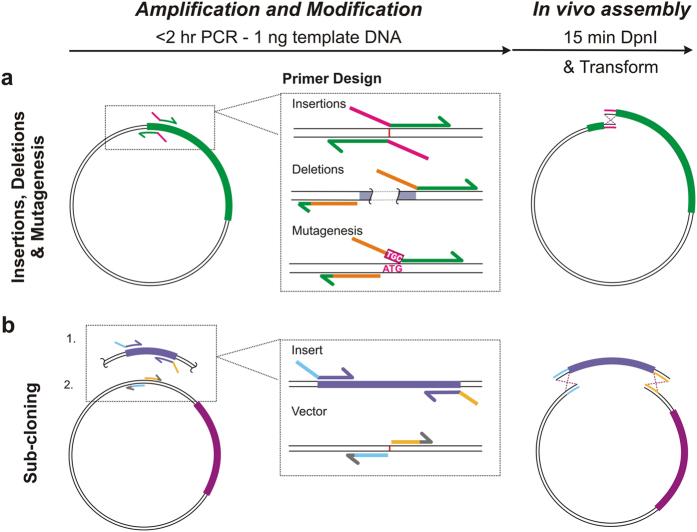
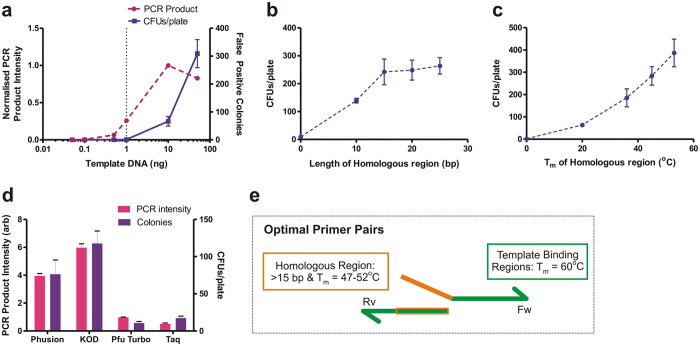
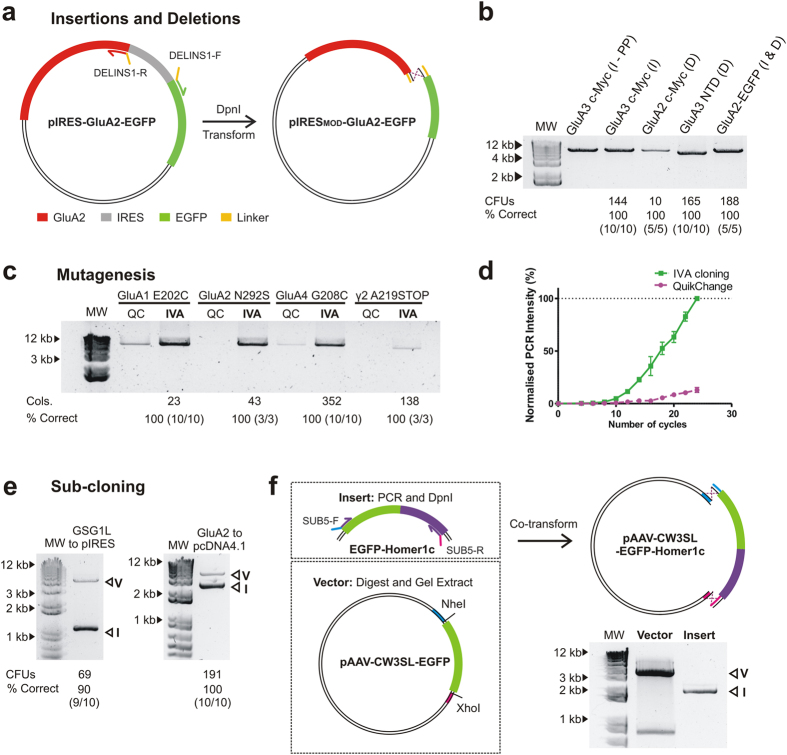
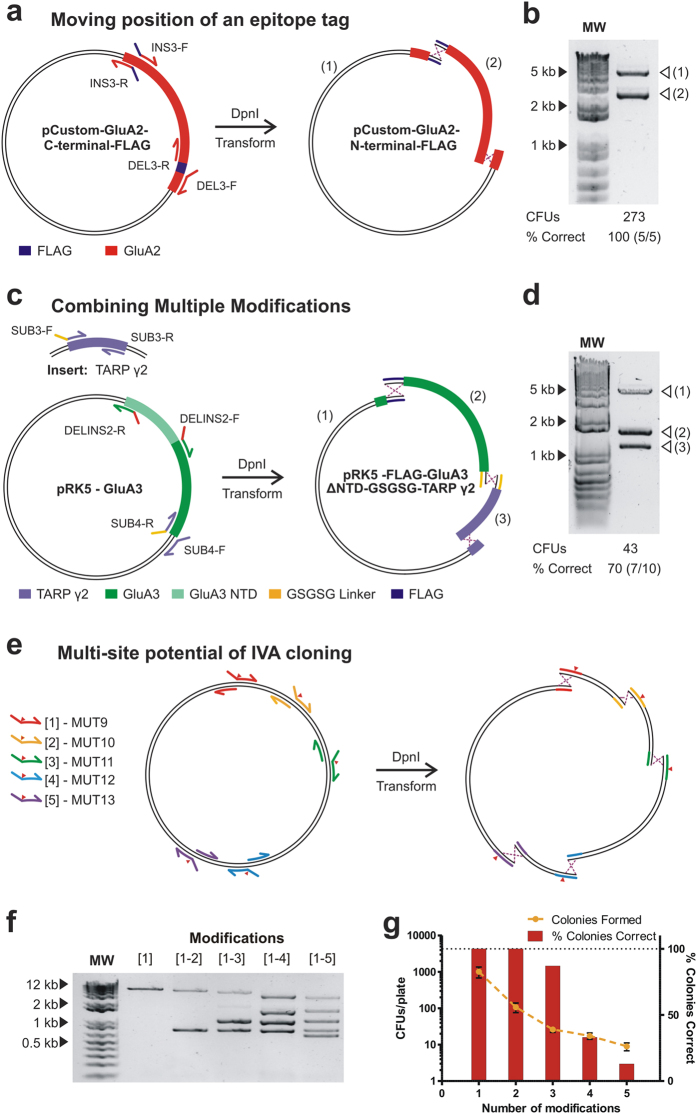
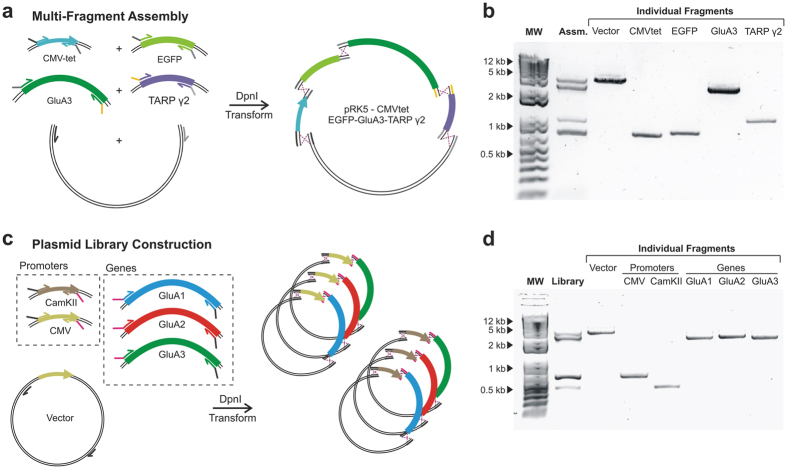
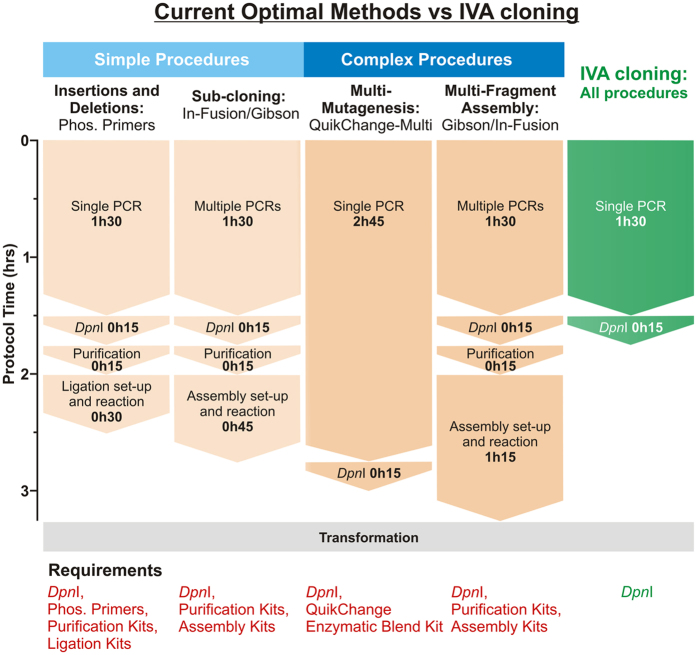
In vivo homologous recombination holds the potential for optimal molecular cloning, however, current strategies require specialised bacterial strains or laborious protocols. Here, we exploit a recA-independent recombination pathway, present in widespread laboratory E.coli strains, to develop IVA (In Vivo Assembly) cloning. This system eliminates the need for enzymatic assembly and reduces all molecular cloning procedures to a single-tube, single-step PCR, performed in <2 hours from setup to transformation. Unlike other methods, IVA is a complete system, and offers significant advantages over alternative methods for all cloning procedures (insertions, deletions, site-directed mutagenesis and sub-cloning). Significantly, IVA allows unprecedented simplification of complex cloning procedures: five simultaneous modifications of any kind, multi-fragment assembly and library construction are performed in approximately half the time of current protocols, still in a single-step fashion. This system is efficient, seamless and sequence-independent, and requires no special kits, enzymes or proprietary bacteria, which will allow its immediate adoption by the academic and industrial molecular biology community.
Figures
References
-
- Green M. R. & Sambrook J. Molecular Cloning: A laboratory manual 4th edn, Vol. 1 Ch. 9, 157–260 (Cold Spring Harbour Laboratory Press, 2012).
-
- Chen B.-Y. & Janes H. W. PCR Cloning Protocols in Methods in Molecular Biology 2nd edn, Vol. 192 (Humana Press, 2002).
-
- Mullis K. et al. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 51, 263–273 (1986). - PubMed
-
- Bryksin A. & Matsamura I. Overlap Extension PCR Cloning. Methods Mol. Biol. 1073, 169–174 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
