

Systematic Targeting of Protein-Protein Interactions
- PMID: 27267699
- PMCID: PMC4961577
- DOI: 10.1016/j.tips.2016.05.008
Systematic Targeting of Protein-Protein Interactions
Abstract
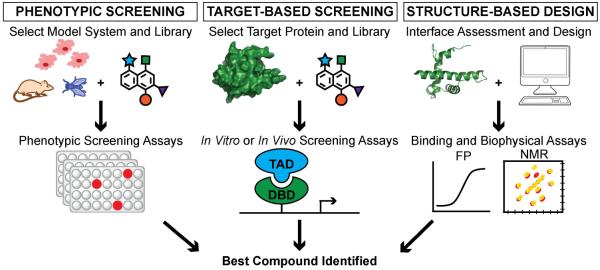
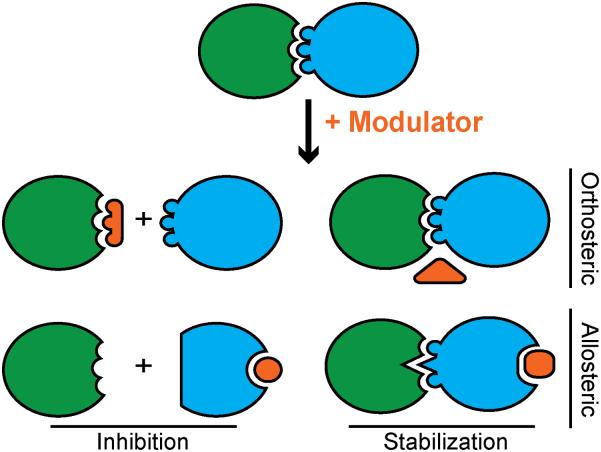
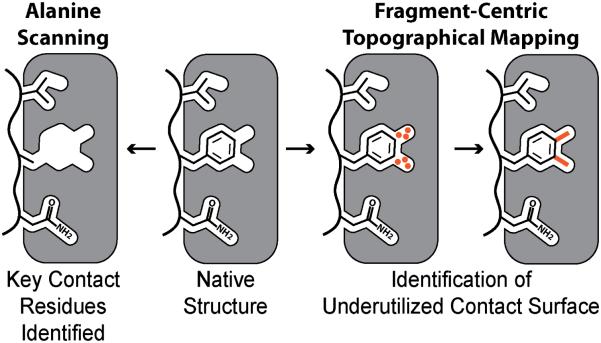
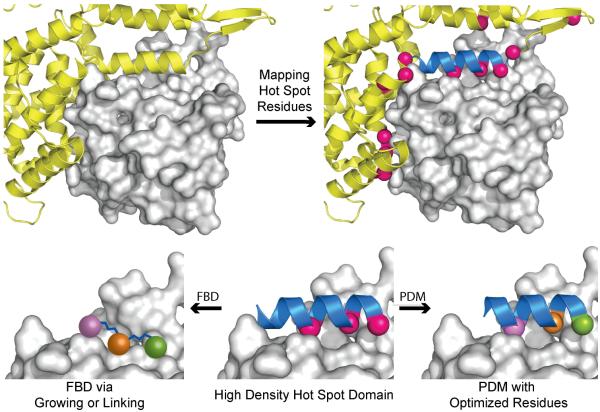
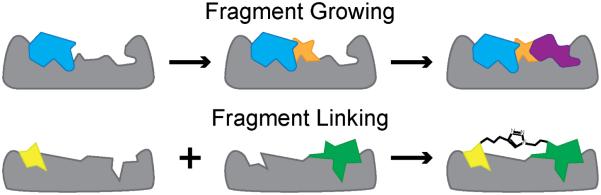
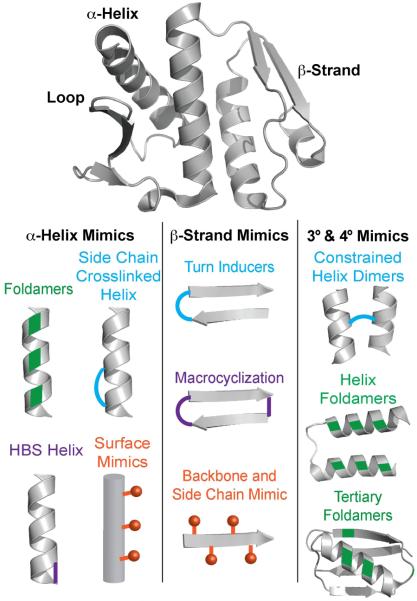
Over the past decade, protein-protein interactions (PPIs) have gone from being neglected as 'undruggable' to being considered attractive targets for the development of therapeutics. Recent advances in computational analysis, fragment-based screening, and molecular design have revealed promising strategies to address the basic molecular recognition challenge: how to target large protein surfaces with specificity. Several systematic and complementary workflows have been developed to yield successful inhibitors of PPIs. Here we review the major contemporary approaches utilized for the discovery of inhibitors and focus on a structure-based workflow, from the selection of a biological target to design.
Keywords: fragment-based design; inhibitors; protein-domain mimics; protein–protein interactions; rational design; screening.
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
References
-
- Swinney DC, Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011:10. - PubMed
-
- Moffat JG, Rudolph J, Bailey D. Phenotypic screening in cancer drug discovery – past, present and future. Nat. Rev. Drug Discov. 2014;13:588–402. - PubMed
-
- Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999;286:971–974. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
