

Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS
- PMID: 27270401
- PMCID: PMC4955684
- DOI: 10.1038/ni.3457
Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS
Abstract
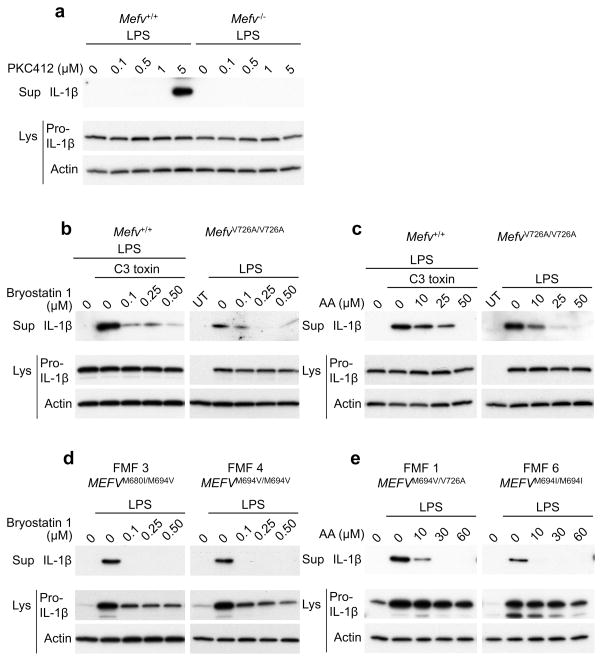
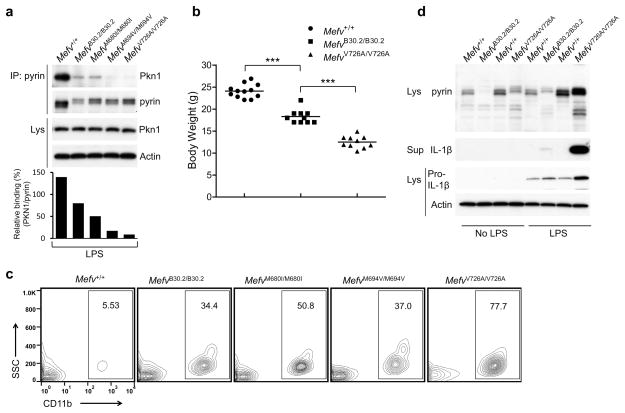
Mutations in the genes encoding pyrin and mevalonate kinase (MVK) cause distinct interleukin-1β (IL-1β)-mediated autoinflammatory diseases: familial Mediterranean fever (FMF) and hyperimmunoglobulinemia D syndrome (HIDS). Pyrin forms an inflammasome when mutant or in response to bacterial modification of the GTPase RhoA. We found that RhoA activated the serine-threonine kinases PKN1 and PKN2 that bind and phosphorylate pyrin. Phosphorylated pyrin bound to 14-3-3 proteins, regulatory proteins that in turn blocked the pyrin inflammasome. The binding of 14-3-3 and PKN proteins to FMF-associated mutant pyrin was substantially decreased, and the constitutive IL-1β release from peripheral blood mononuclear cells of patients with FMF or HIDS was attenuated by activation of PKN1 and PKN2. Defects in prenylation, seen in HIDS, led to RhoA inactivation and consequent pyrin inflammasome activation. These data suggest a previously unsuspected fundamental molecular connection between two seemingly distinct autoinflammatory disorders.
Conflict of interest statement
The authors declare no competing financial interests.
Figures






Comment in
-
A dRAStic RHOAdblock of Pyrin inflammasome activation.Nat Immunol. 2016 Jul 19;17(8):900-2. doi: 10.1038/ni.3511. Nat Immunol. 2016. PMID: 27434003 Free PMC article.
References
-
- International_FMF_Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90:797–807. - PubMed
-
- Drenth JP, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet. 1999;22:178–181. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
