

Base changes in tumour DNA have the power to reveal the causes and evolution of cancer
- PMID: 27270430
- PMCID: PMC5241425
- DOI: 10.1038/onc.2016.192
Base changes in tumour DNA have the power to reveal the causes and evolution of cancer
Abstract
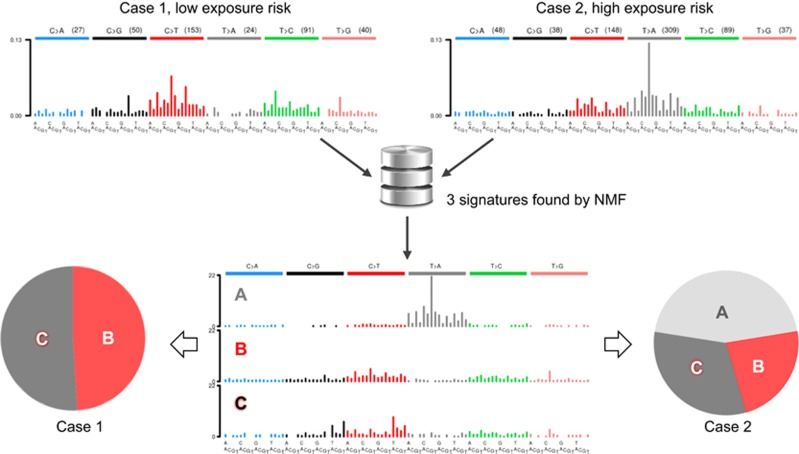
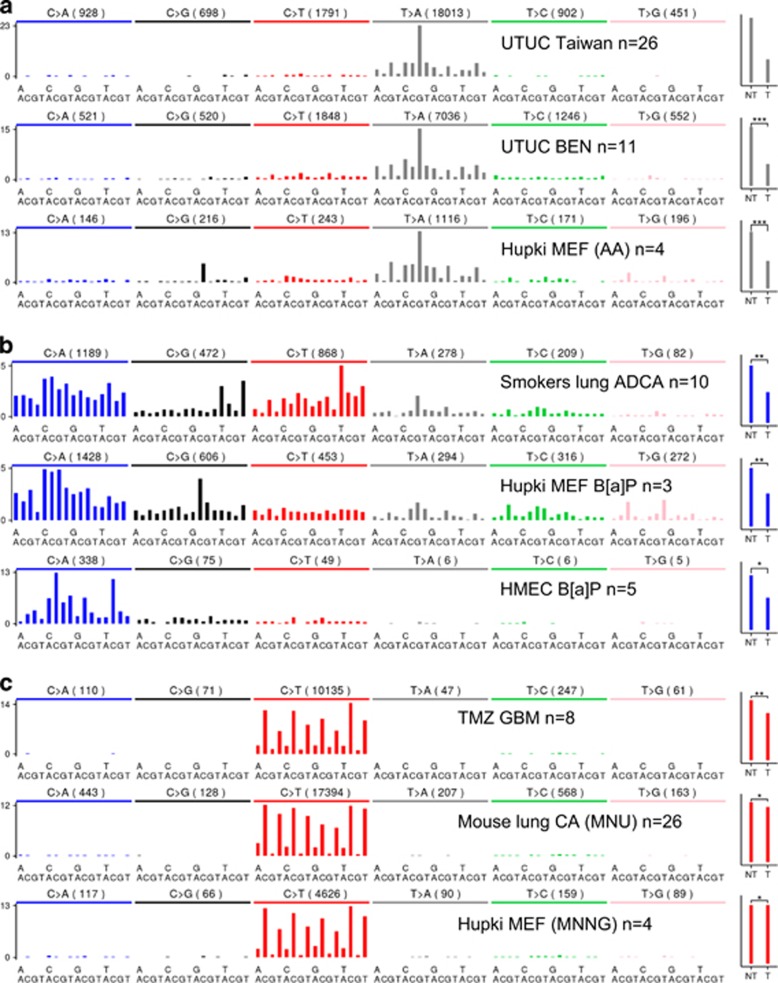
Next-generation sequencing (NGS) technology has demonstrated that the cancer genomes are peppered with mutations. Although most somatic tumour mutations are unlikely to have any role in the cancer process per se, the spectra of DNA sequence changes in tumour mutation catalogues have the potential to identify the mutagens, and to reveal the mutagenic processes responsible for human cancer. Very recently, a novel approach for data mining of the vast compilations of tumour NGS data succeeded in separating and precisely defining at least 30 distinct patterns of sequence change hidden in mutation databases. At least half of these mutational signatures can be readily assigned to known human carcinogenic exposures or endogenous mechanisms of mutagenesis. A quantum leap in our knowledge of mutagenesis in human cancers has resulted, stimulating a flurry of research activity. We trace here the major findings leading first to the hypothesis that carcinogenic insults leave characteristic imprints on the DNA sequence of tumours, and culminating in empirical evidence from NGS data that well-defined carcinogen mutational signatures are indeed present in tumour genomic DNA from a variety of cancer types. The notion that tumour DNAs can divulge environmental sources of mutation is now a well-accepted fact. This approach to cancer aetiology has also incriminated various endogenous, enzyme-driven processes that increase the somatic mutation load in sporadic cancers. The tasks now confronting the field of molecular epidemiology are to assign mutagenic processes to orphan and newly discovered tumour mutation patterns, and to determine whether avoidable cancer risk factors influence signatures produced by endogenous enzymatic mechanisms. Innovative research with experimental models and exploitation of the geographical heterogeneity in cancer incidence can address these challenges.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
