

SCN8A encephalopathy: Research progress and prospects
- PMID: 27270488
- PMCID: PMC5495462
- DOI: 10.1111/epi.13422
SCN8A encephalopathy: Research progress and prospects
Abstract
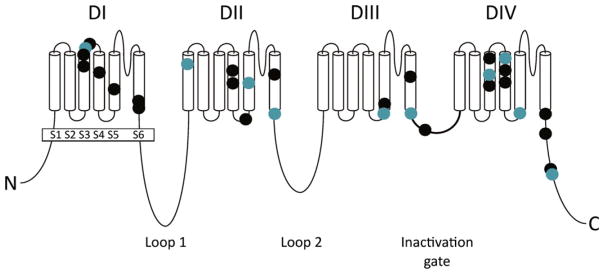
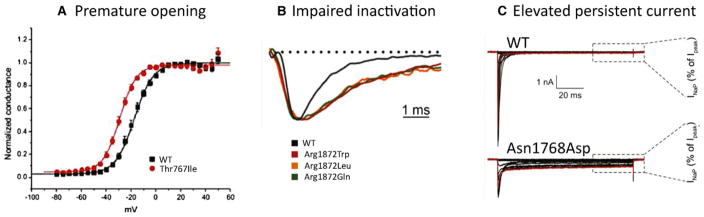
On April 21, 2015, the first SCN8A Encephalopathy Research Group convened in Washington, DC, to assess current research into clinical and pathogenic features of the disorder and prepare an agenda for future research collaborations. The group comprised clinical and basic scientists and representatives of patient advocacy groups. SCN8A encephalopathy is a rare disorder caused by de novo missense mutations of the sodium channel gene SCN8A, which encodes the neuronal sodium channel Nav 1.6. Since the initial description in 2012, approximately 140 affected individuals have been reported in publications or by SCN8A family groups. As a result, an understanding of the severe impact of SCN8A mutations is beginning to emerge. Defining a genetic epilepsy syndrome goes beyond identification of molecular etiology. Topics discussed at this meeting included (1) comparison between mutations of SCN8A and the SCN1A mutations in Dravet syndrome, (2) biophysical properties of the Nav 1.6 channel, (3) electrophysiologic effects of patient mutations on channel properties, (4) cell and animal models of SCN8A encephalopathy, (5) drug screening strategies, (6) the phenotypic spectrum of SCN8A encephalopathy, and (7) efforts to develop a bioregistry. A panel discussion of gaps in bioregistry, biobanking, and clinical outcomes data was followed by a planning session for improved integration of clinical and basic science research. Although SCN8A encephalopathy was identified only recently, there has been rapid progress in functional analysis and phenotypic classification. The focus is now shifting from identification of the underlying molecular cause to the development of strategies for drug screening and prioritized patient care.
Keywords: Bioregistry; Drug screening; Encephalopathy; Mutation; Nav1.6; SCN8A; Sodium channel.
Wiley Periodicals, Inc. © 2016 International League Against Epilepsy.
Figures

References
-
- Russ SA, Larson K, Halfon N. A national profile of childhood epilepsy and seizure disorder. Pediatrics. 2012;129:256–264. - PubMed
-
- Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
