

Directed vaccination against pneumococcal disease
- PMID: 27274071
- PMCID: PMC4922154
- DOI: 10.1073/pnas.1603007113
Directed vaccination against pneumococcal disease
Abstract
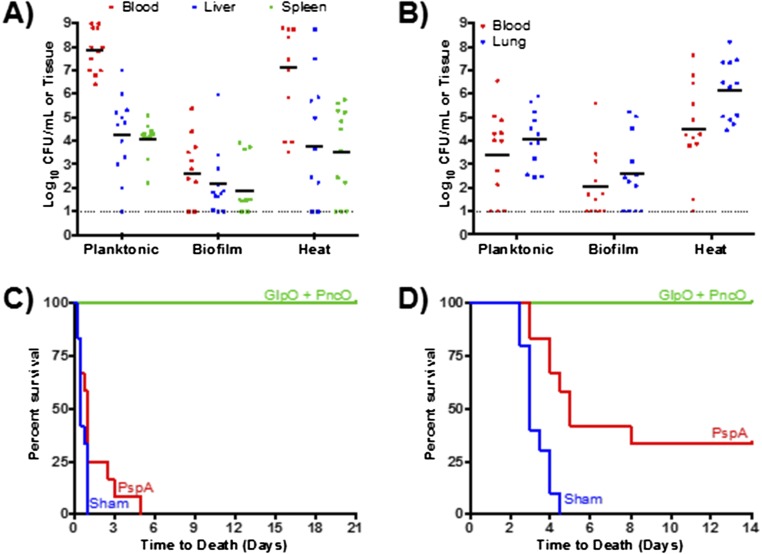
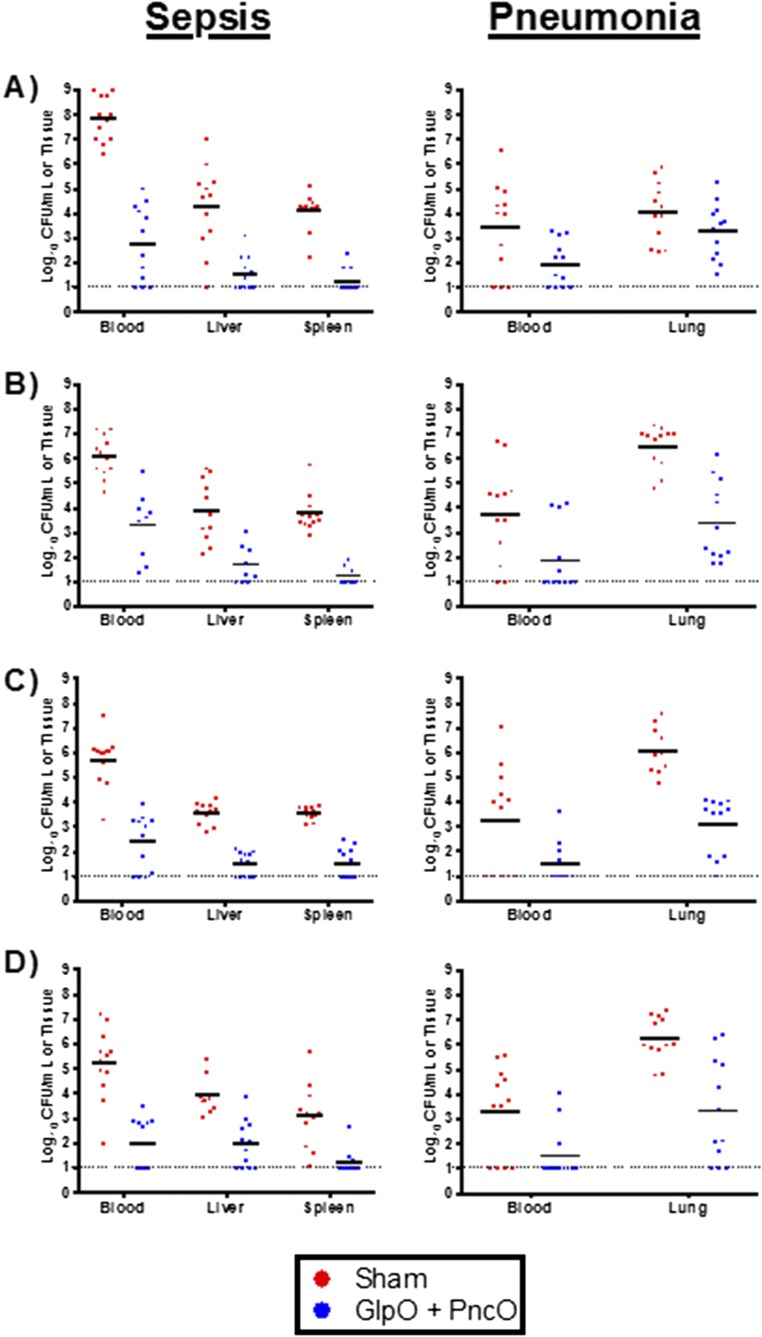
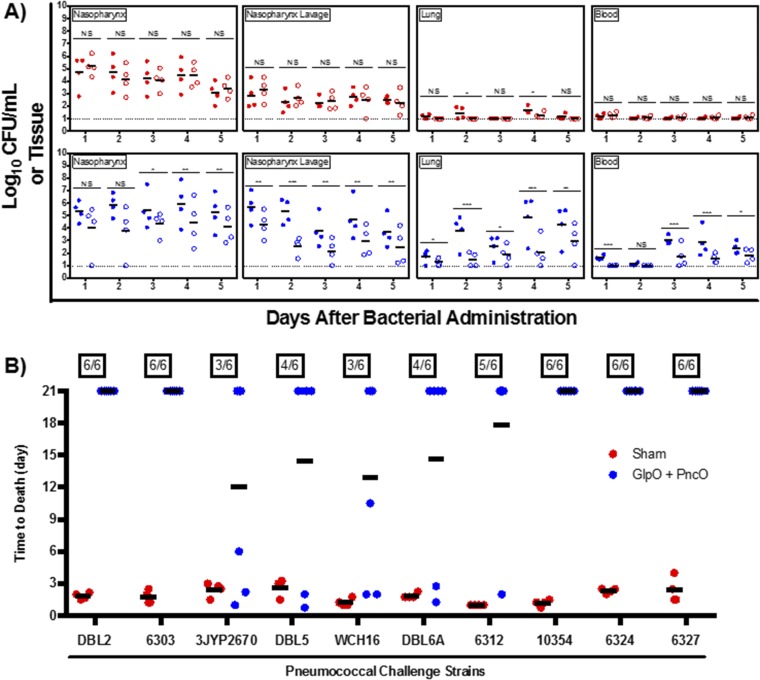
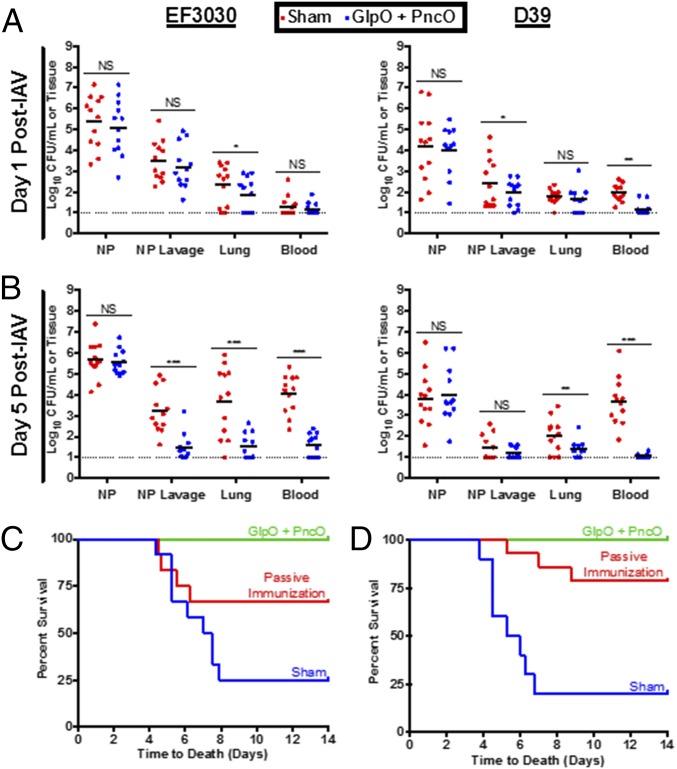
Immunization strategies against commensal bacterial pathogens have long focused on eradicating asymptomatic carriage as well as disease, resulting in changes in the colonizing microflora with unknown future consequences. Additionally, current vaccines are not easily adaptable to sequence diversity and immune evasion. Here, we present a "smart" vaccine that leverages our current understanding of disease transition from bacterial carriage to infection with the pneumococcus serving as a model organism. Using conserved surface proteins highly expressed during virulent transition, the vaccine mounts an immune response specifically against disease-causing bacterial populations without affecting carriage. Aided by a delivery technology capable of multivalent surface display, which can be adapted easily to a changing clinical picture, results include complete protection against the development of pneumonia and sepsis during animal challenge experiments with multiple, highly variable, and clinically relevant pneumococcal isolates. The approach thus offers a unique and dynamic treatment option readily adaptable to other commensal pathogens.
Keywords: S. pneumoniae; biofilm; pneumococcal; vaccine; virulence.
Conflict of interest statement
The authors declare no conflict of interest.
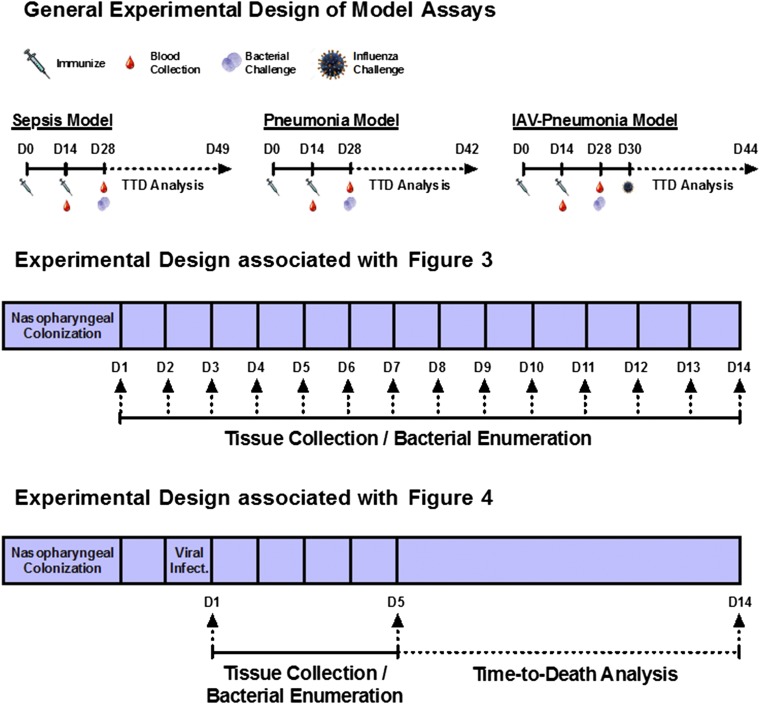
Figures
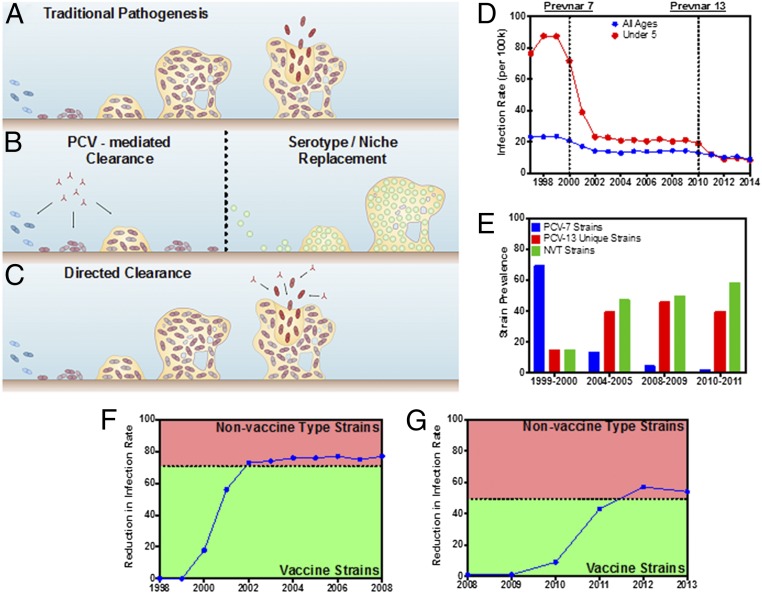
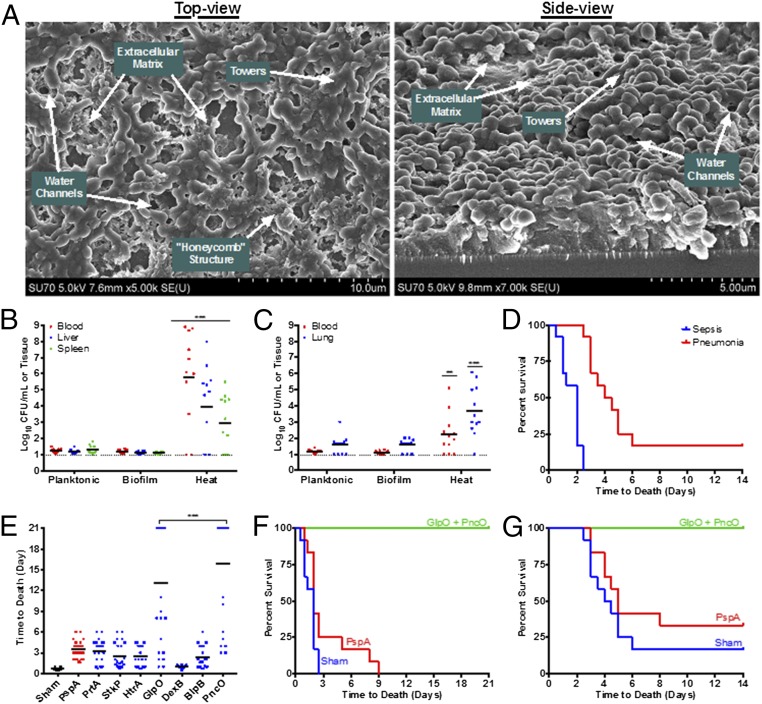
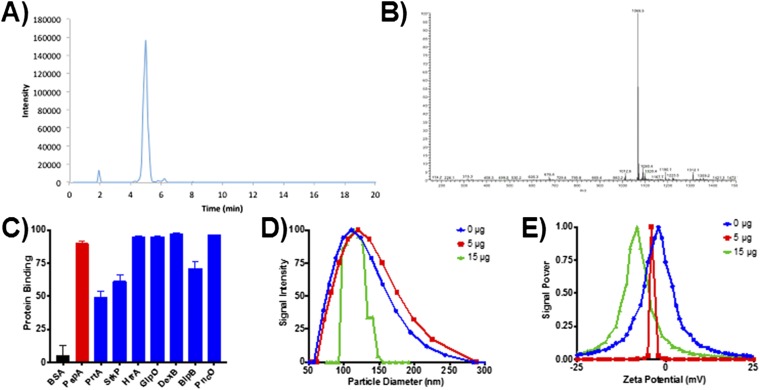
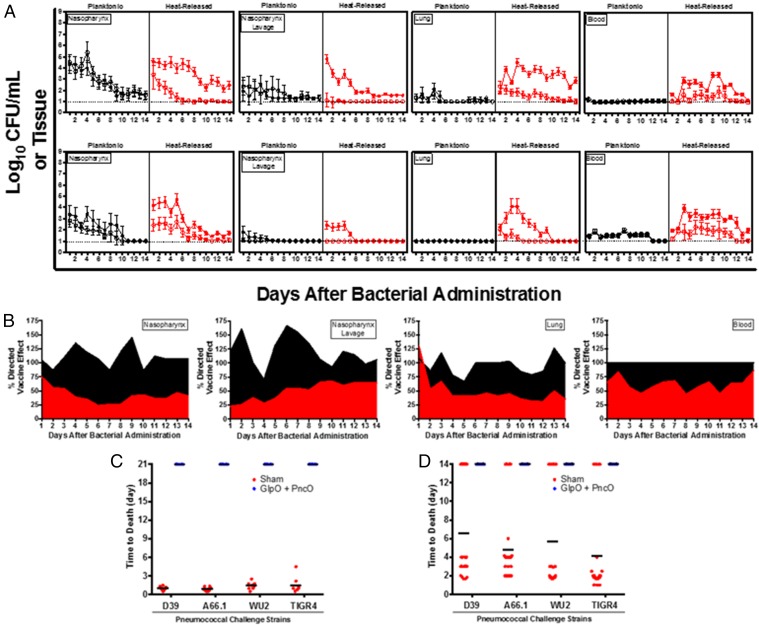
References
-
- Hausdorff WP, Hoet B, Adegbola RA. Predicting the impact of new pneumococcal conjugate vaccines: Serotype composition is not enough. Expert Rev Vaccines. 2015;14(3):413–428. - PubMed
-
- Bogaert D, De Groot R, Hermans PWM. Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect Dis. 2004;4(3):144–154. - PubMed
-
- Gray BM, Converse GM, 3rd, Dillon HC., Jr Epidemiologic studies of Streptococcus pneumoniae in infants: Acquisition, carriage, and infection during the first 24 months of life. J Infect Dis. 1980;142(6):923–933. - PubMed
-
- Huebner RE, Dagan R, Porath N, Wasas AD, Klugman KP. Lack of utility of serotyping multiple colonies for detection of simultaneous nasopharyngeal carriage of different pneumococcal serotypes. Pediatr Infect Dis J. 2000;19(10):1017–1020. - PubMed
-
- Heron MP, Smith BL. Deaths: Leading causes for 2003. National vital statistics reports: From the Centers for Disease Control and Prevention, National Center for Health Statistics. National Vital Statistics System. 2007;55(10):1–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
