

Mutation of TBCK causes a rare recessive developmental disorder
- PMID: 27275012
- PMCID: PMC4881620
- DOI: 10.1212/NXG.0000000000000076
Mutation of TBCK causes a rare recessive developmental disorder
Erratum in
-
Erratum: Mutation of TBCK causes a rare recessive developmental disorder.Neurol Genet. 2016 Aug 4;2(4):e86. doi: 10.1212/NXG.0000000000000086. eCollection 2016 Aug. Neurol Genet. 2016. PMID: 29441365 Free PMC article.
Abstract
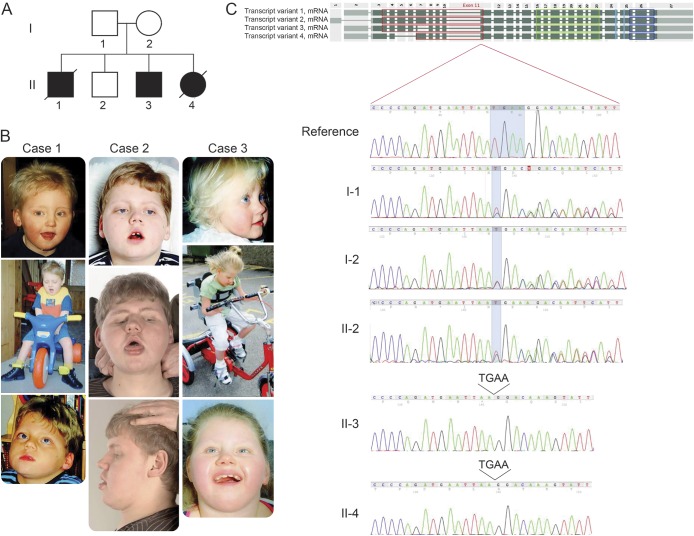
Objective: To characterize the underlying genetic defect in a family with 3 siblings affected by a severe, yet viable, congenital disorder.
Methods: Extensive genetic and metabolic investigations were performed, and the affected children were imaged at different ages. Whole-genome genotyping and whole-exome sequencing were undertaken. A single large region (>8 Mb) of homozygosity in chromosome 4 (chr4:100,268,553-108,609,628) was identified that was shared only in affected siblings. Inspection of genetic variability within this region led to the identification of a novel mutation. Sanger sequencing confirmed segregation of the mutation with disease.
Results: All affected siblings share homozygosity for a novel 4-bp deletion in the gene TBCK (NM_033115:c.614_617del:p.205_206del).
Conclusions: This finding provides the genetic cause of a severe inherited disease in a family and extends the number of mutations and phenotypes associated with this recently identified disease gene.
Figures
References
-
- Bras J, Guerreiro R, Hardy J. Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev 2012;13:453–464. - PubMed
-
- Smith A, Leask K, Tomlin P, Donnai D. A familial dysmorphic condition with hypotonia, seizures and precocious puberty. Clin Dysmorphol 2008;17:161–164. - PubMed
-
- Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened mulitplex consanguineous families. Cell Rep 2015;10:148–161. - PubMed
-
- Collinet C, Stöter M, Bradshaw CR, et al. Systems survey of endocytosis by multiparametric image analysis. Nature 2010;464:243–249. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous