

Characterising the Canine Oral Microbiome by Direct Sequencing of Reverse-Transcribed rRNA Molecules
- PMID: 27276347
- PMCID: PMC4898712
- DOI: 10.1371/journal.pone.0157046
Characterising the Canine Oral Microbiome by Direct Sequencing of Reverse-Transcribed rRNA Molecules
Abstract
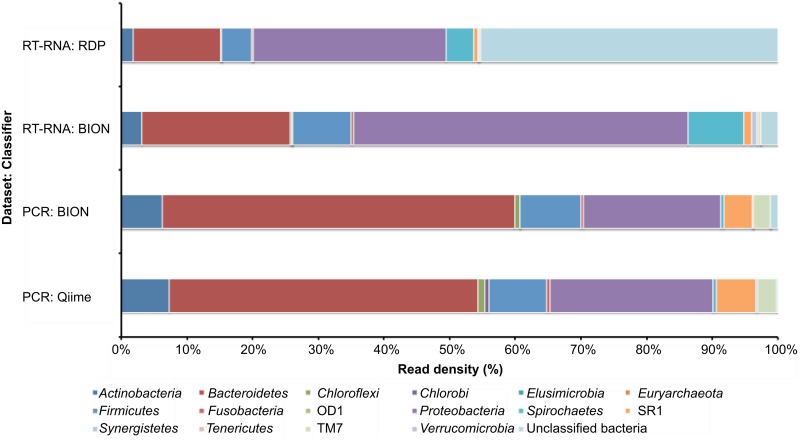
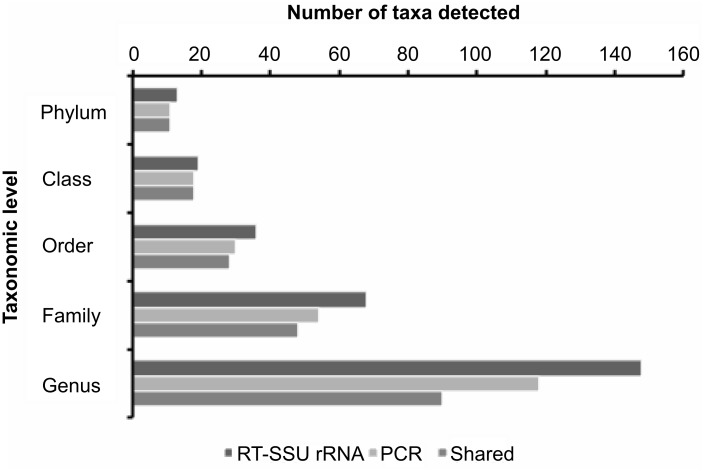
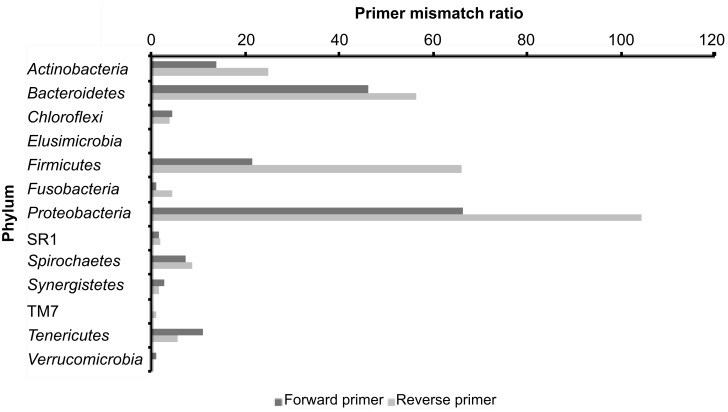
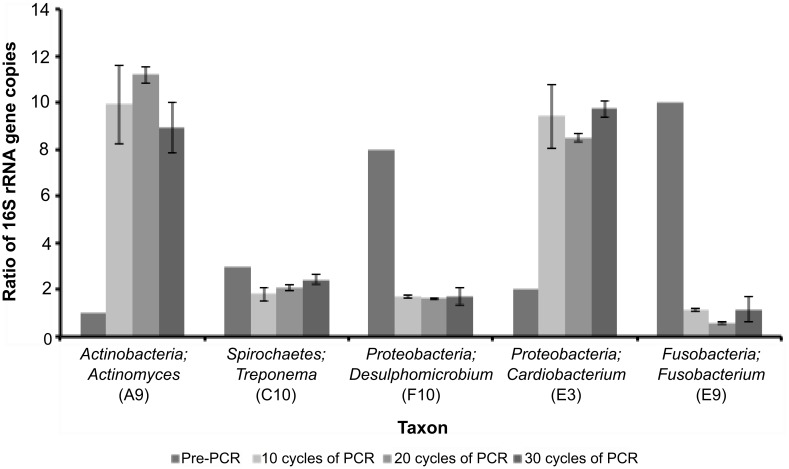
PCR amplification and sequencing of phylogenetic markers, primarily Small Sub-Unit ribosomal RNA (SSU rRNA) genes, has been the paradigm for defining the taxonomic composition of microbiomes. However, 'universal' SSU rRNA gene PCR primer sets are likely to miss much of the diversity therein. We sequenced a library comprising purified and reverse-transcribed SSU rRNA (RT-SSU rRNA) molecules from the canine oral microbiome and compared it to a general bacterial 16S rRNA gene PCR amplicon library generated from the same biological sample. In addition, we have developed BIONmeta, a novel, open-source, computer package for the processing and taxonomic classification of the randomly fragmented RT-SSU rRNA reads produced. Direct RT-SSU rRNA sequencing revealed that 16S rRNA molecules belonging to the bacterial phyla Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria and Spirochaetes, were most abundant in the canine oral microbiome (92.5% of total bacterial SSU rRNA). The direct rRNA sequencing approach detected greater taxonomic diversity (1 additional phylum, 2 classes, 1 order, 10 families and 61 genera) when compared with general bacterial 16S rRNA amplicons from the same sample, simultaneously provided SSU rRNA gene inventories of Bacteria, Archaea and Eukarya, and detected significant numbers of sequences not recognised by 'universal' primer sets. Proteobacteria and Spirochaetes were found to be under-represented by PCR-based analysis of the microbiome, and this was due to primer mismatches and taxon-specific variations in amplification efficiency, validated by qPCR analysis of 16S rRNA amplicons from a mock community. This demonstrated the veracity of direct RT-SSU rRNA sequencing for molecular microbial ecology.
Conflict of interest statement
Figures

References
-
- Pace NR. A molecular view of microbial diversity and the biosphere. Science. 1997;276: 734–740. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
