

Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons
- PMID: 27277231
- PMCID: PMC4899909
- DOI: 10.1186/s13024-016-0111-6
Stathmin 1/2-triggered microtubule loss mediates Golgi fragmentation in mutant SOD1 motor neurons
Abstract
Background: Pathological Golgi fragmentation represents a constant pre-clinical feature of many neurodegenerative diseases including amyotrophic lateral sclerosis (ALS) but its molecular mechanisms remain hitherto unclear.
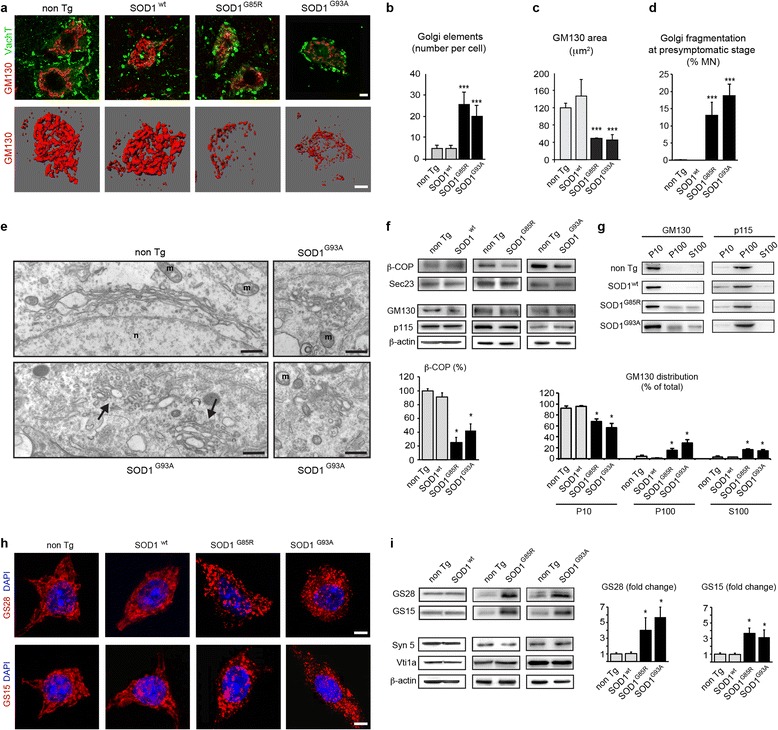
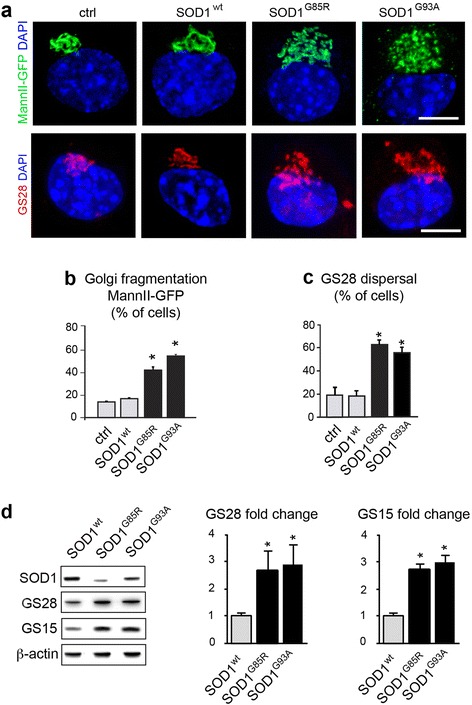
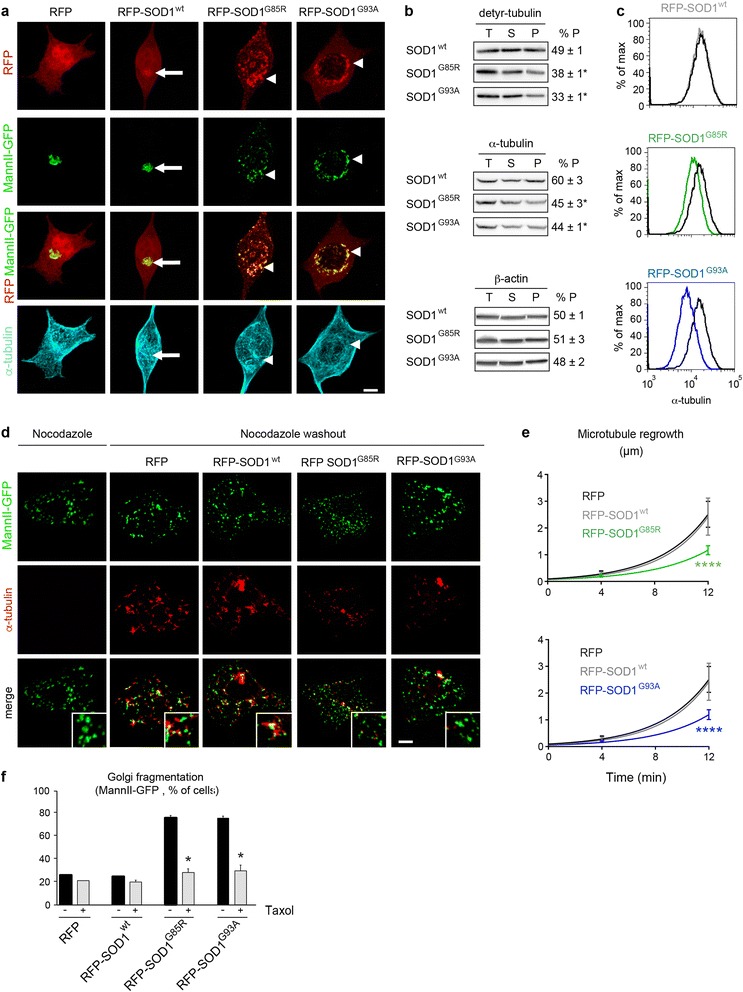
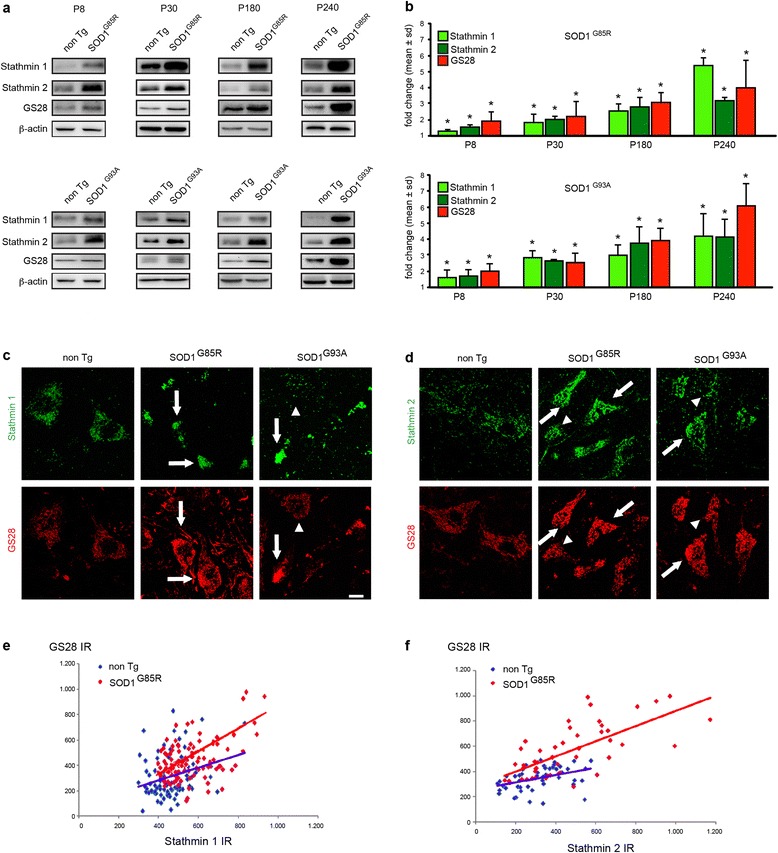
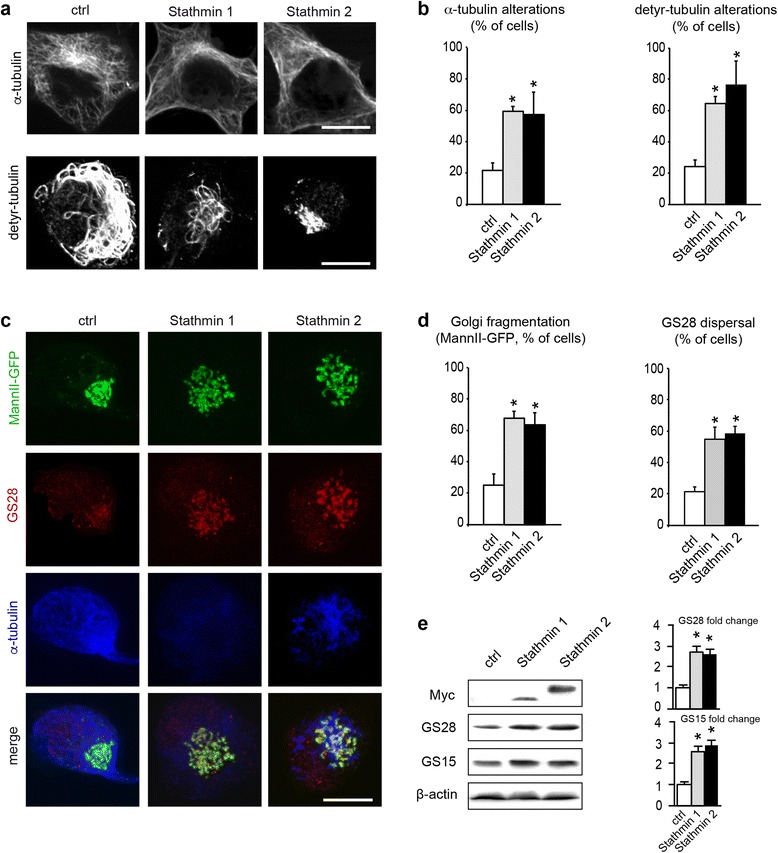
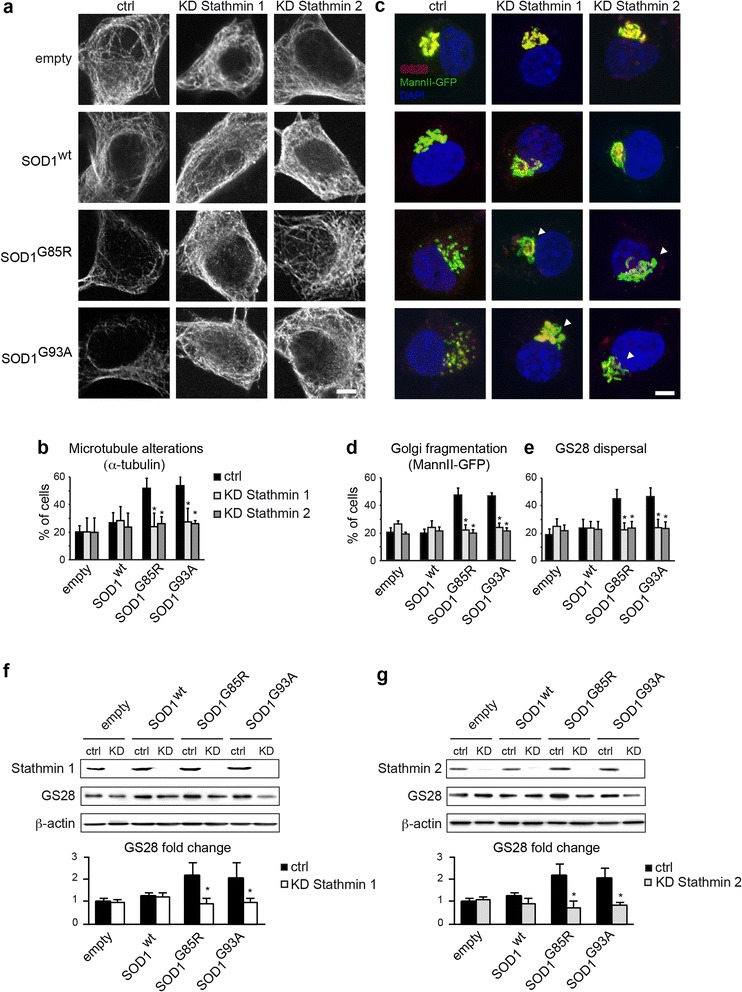
Results: Here, we show that the severe Golgi fragmentation in transgenic mutant SOD1(G85R) and SOD1(G93A) mouse motor neurons is associated with defective polymerization of Golgi-derived microtubules, loss of the COPI coat subunit β-COP, cytoplasmic dispersion of the Golgi tether GM130, strong accumulation of the ER-Golgi v-SNAREs GS15 and GS28 as well as tubular/vesicular Golgi fragmentation. Data mining, transcriptomic and protein analyses demonstrate that both SOD1 mutants cause early presymptomatic and rapidly progressive up-regulation of the microtubule-destabilizing proteins Stathmins 1 and 2. Remarkably, mutant SOD1-triggered Golgi fragmentation and Golgi SNARE accumulation are recapitulated by Stathmin 1/2 overexpression but completely rescued by Stathmin 1/2 knockdown or the microtubule-stabilizing drug Taxol.
Conclusions: We conclude that Stathmin-triggered microtubule destabilization mediates Golgi fragmentation in mutant SOD1-linked ALS and potentially also in related motor neuron diseases.
Figures
References
-
- Acevedo-Arozena A, Kalmar B, Essa S, Ricketts T, Joyce P, Kent R, Rowe C, Parker A, Gray A, Hafezparast M, et al. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis model Mech. 2011;4:686–700. doi: 10.1242/dmm.007237. - DOI - PMC - PubMed
-
- Aguilera-Gomez A, Rabouille C. Intra Golgi transport. In: Bradshaw R, Stahl P (eds) Encyclopedia of Cell Biology. Elsevier; 2015; 354-362. doi:10.1016/B978-0-12-394447-4.20034-5.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
