

Defining the spectrum of frontotemporal dementias associated with TARDBP mutations
- PMID: 27280171
- PMCID: PMC4882769
- DOI: 10.1212/NXG.0000000000000080
Defining the spectrum of frontotemporal dementias associated with TARDBP mutations
Abstract
Objectives: We describe the largest series of patients with TARDBP mutations presenting with frontotemporal dementia (FTD) and review the cases in the literature to precisely characterize FTD diseases associated with this genotype.
Methods: The phenotypic characteristics of 29 TARDBP patients, including 10 new French and Dutch cases and 19 reviewed from the literature, were evaluated.
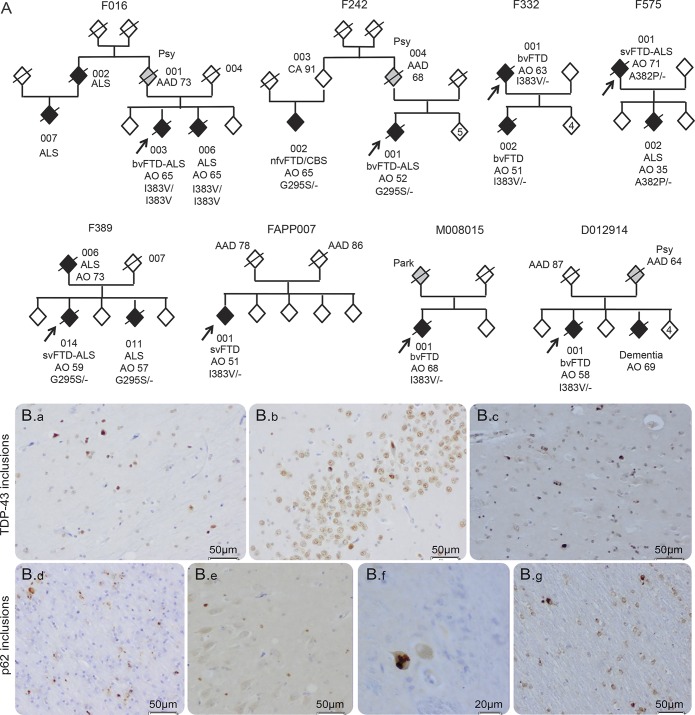
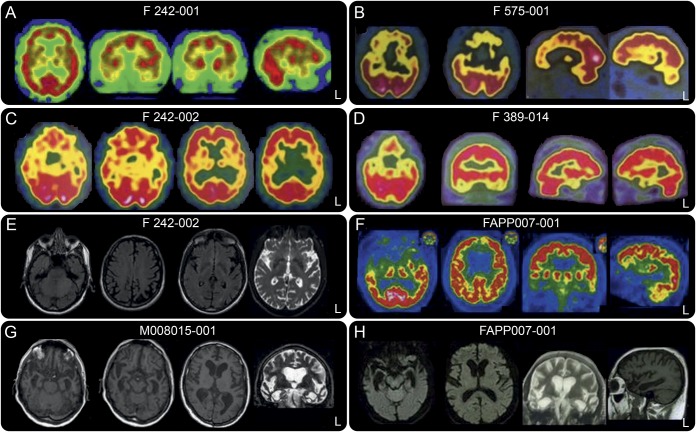
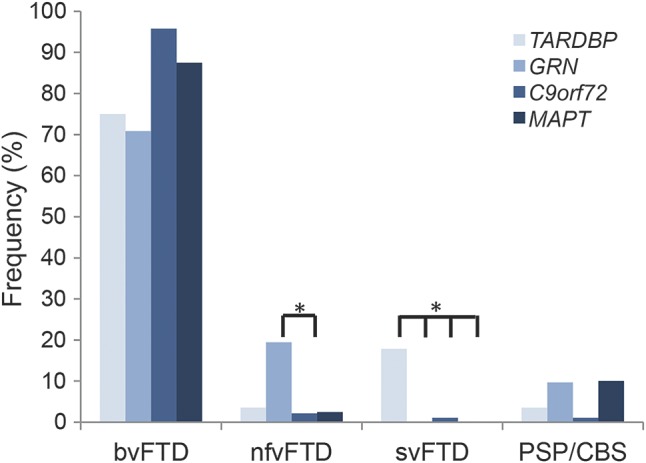
Results: The most frequent phenotype was a behavioral variant frontotemporal dementia (bvFTD), but a significant proportion (40%) of our patients had semantic (svFTD) or nonfluent variants (nfvFTD) at onset; and svFTD was significantly more frequent in TARDBP carriers than in other FTD genotypes (p < 0.001). Remarkably, only a minority (40%) of our patients secondarily developed amyotrophic lateral sclerosis (ALS). Two patients carried a homozygous mutation but strikingly different phenotypes (bvFTD and ALS) indicating that homozygosity does not result in a specific phenotype. Earlier age at onset in children than parent's generations, mimicking an apparent "anticipation" (21.8 ± 9.3 years, p = 0.001), and possible reduced penetrance were present in most families.
Conclusions: This study enlarges the phenotypic spectrum of TARDBP and will have important clinical implications: (1) FTD can be the only clinical manifestation of TARDBP mutations; (2) Initial language or semantic disorders might be indicative of a specific genotype; (3) Mutations should be searched in all FTD phenotypes after exclusion of major genes, even in the absence of ALS in the proband or in family history; (4) reduced penetrance and clinical variability should be considered to deliver appropriate genetic counseling.
Figures
References
-
- Lattante S, Rouleau GA, Kabashi E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum Mutat 2013;34:812–826. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous