

Distinct Roles for Intracellular and Extracellular Lipids in Hepatitis C Virus Infection
- PMID: 27280294
- PMCID: PMC4900644
- DOI: 10.1371/journal.pone.0156996
Distinct Roles for Intracellular and Extracellular Lipids in Hepatitis C Virus Infection
Abstract
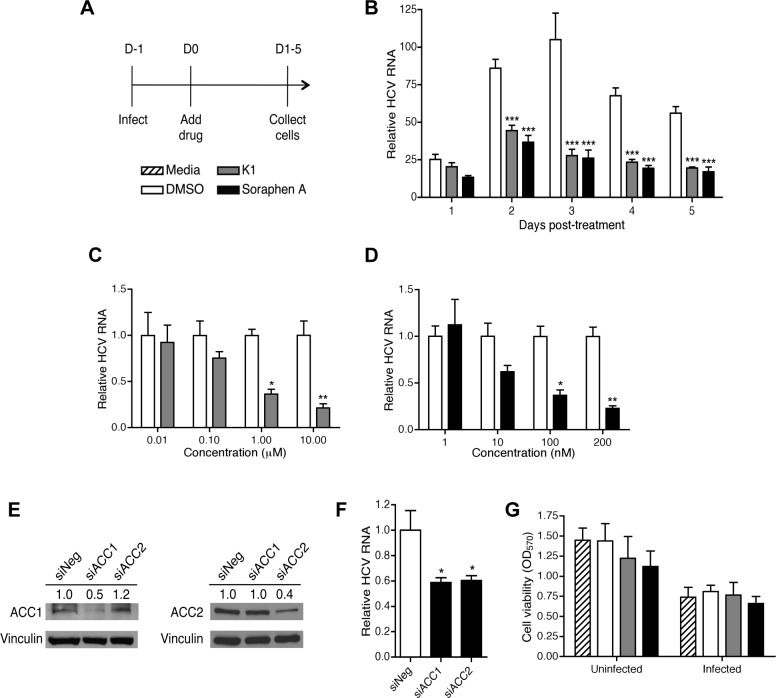
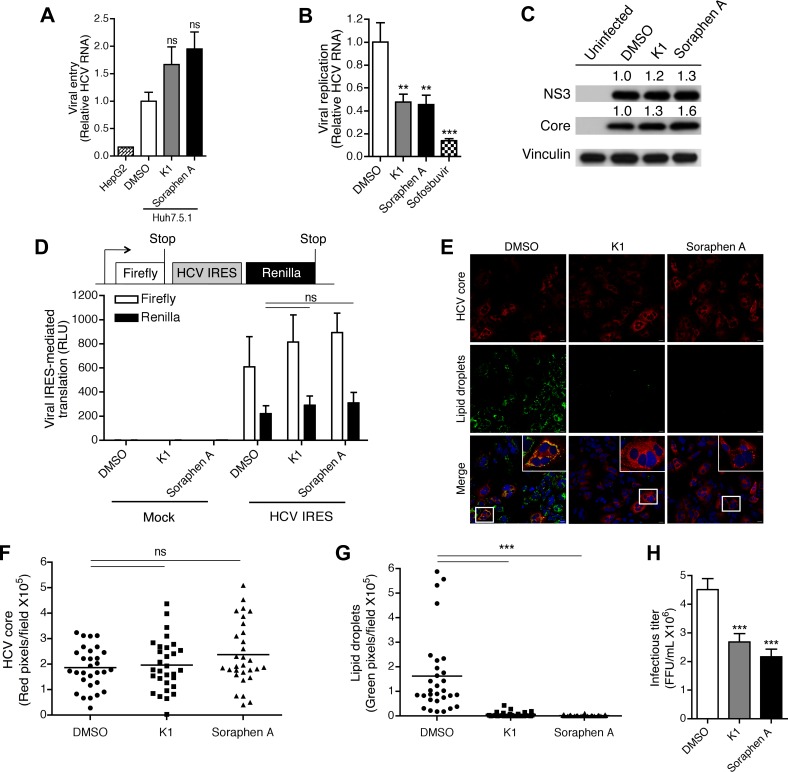
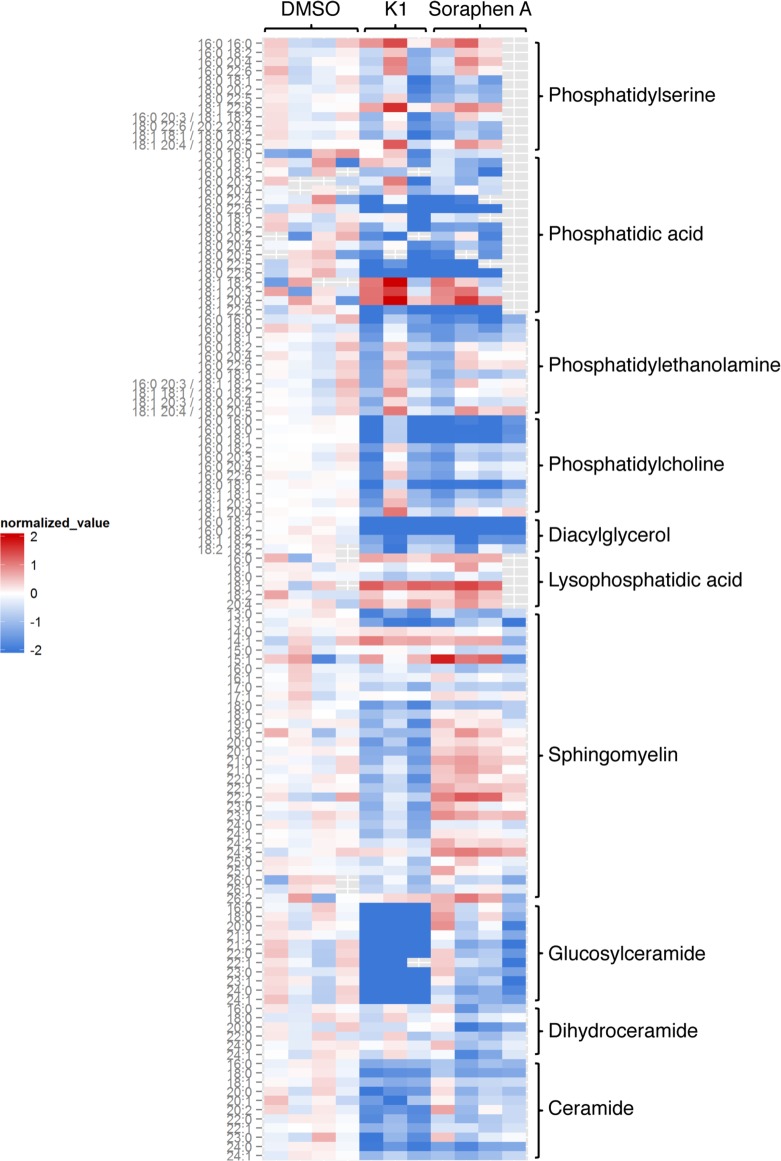
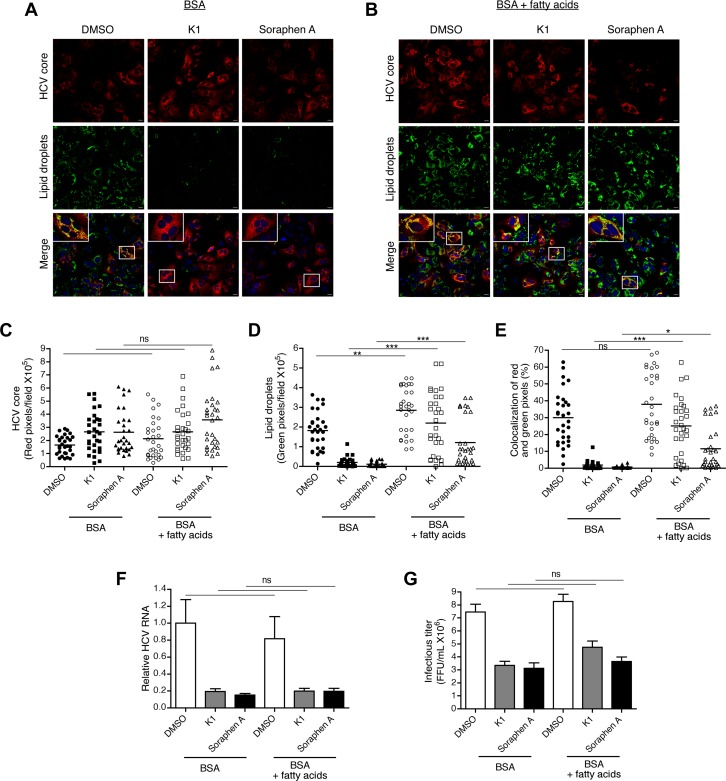
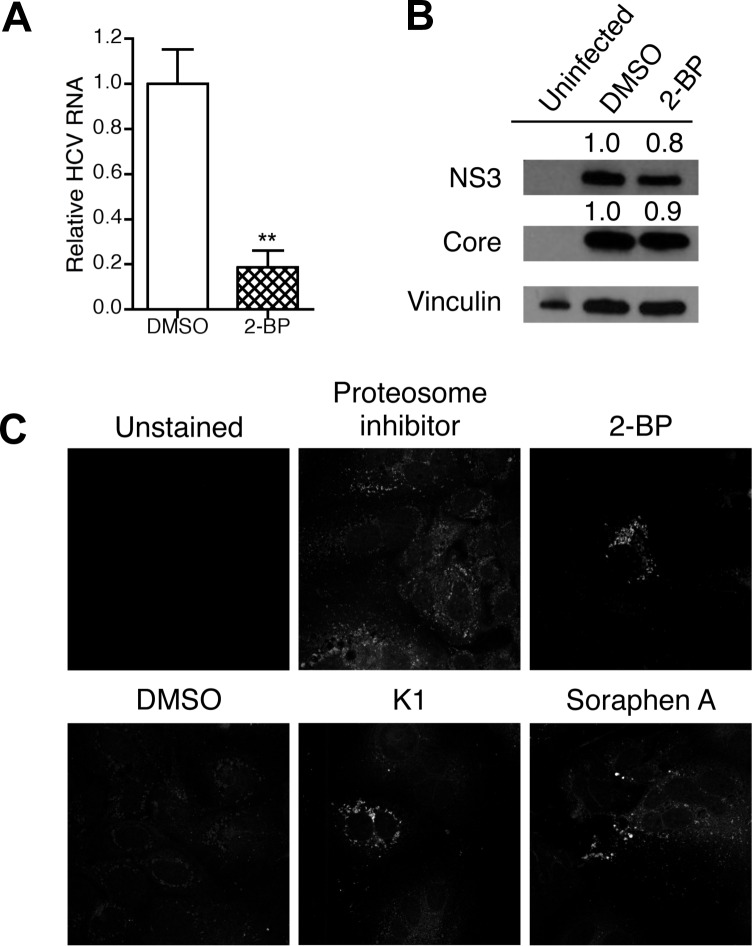
Hepatitis C is a chronic liver disease that contributes to progressive metabolic dysfunction. Infection of hepatocytes by hepatitis C virus (HCV) results in reprogramming of hepatic and serum lipids. However, the specific contribution of these distinct pools of lipids to HCV infection remains ill defined. In this study, we investigated the role of hepatic lipogenesis in HCV infection by targeting the rate-limiting step in this pathway, which is catalyzed by the acetyl-CoA carboxylase (ACC) enzymes. Using two structurally unrelated ACC inhibitors, we determined that blockade of lipogenesis resulted in reduced viral replication, assembly, and release. Supplementing exogenous lipids to cells treated with ACC inhibitors rescued HCV assembly with no effect on viral replication and release. Intriguingly, loss of viral RNA was not recapitulated at the protein level and addition of 2-bromopalmitate, a competitive inhibitor of protein palmitoylation, mirrored the effects of ACC inhibitors on reduced viral RNA without a concurrent loss in protein expression. These correlative results suggest that newly synthesized lipids may have a role in protein palmitoylation during HCV infection.
Conflict of interest statement
Figures
Similar articles
-
Whole Lotta Lipids-from HCV RNA Replication to the Mature Viral Particle.Int J Mol Sci. 2020 Apr 21;21(8):2888. doi: 10.3390/ijms21082888. Int J Mol Sci. 2020. PMID: 32326151 Free PMC article. Review.
-
Forkhead box transcription factor regulation and lipid accumulation by hepatitis C virus.J Virol. 2014 Apr;88(8):4195-203. doi: 10.1128/JVI.03327-13. Epub 2014 Jan 29. J Virol. 2014. PMID: 24478438 Free PMC article.
-
Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis C treatment.Antiviral Res. 2016 Aug;132:262-7. doi: 10.1016/j.antiviral.2016.07.003. Epub 2016 Jul 5. Antiviral Res. 2016. PMID: 27392483
-
Perturbation of de novo lipogenesis hinders MERS-CoV assembly and release, but not the biogenesis of viral replication organelles.J Virol. 2025 Mar 18;99(3):e0228224. doi: 10.1128/jvi.02282-24. Epub 2025 Feb 20. J Virol. 2025. PMID: 39976449 Free PMC article.
-
Roles of phosphoinositides and phosphoinositides kinases in hepatitis C virus RNA replication.Arch Pharm Res. 2012 Oct;35(10):1701-11. doi: 10.1007/s12272-012-1001-2. Epub 2012 Nov 9. Arch Pharm Res. 2012. PMID: 23139120 Review.
Cited by
-
Hijacking the Supplies: Metabolism as a Novel Facet of Virus-Host Interaction.Front Immunol. 2019 Jul 3;10:1533. doi: 10.3389/fimmu.2019.01533. eCollection 2019. Front Immunol. 2019. PMID: 31333664 Free PMC article. Review.
-
Whole Lotta Lipids-from HCV RNA Replication to the Mature Viral Particle.Int J Mol Sci. 2020 Apr 21;21(8):2888. doi: 10.3390/ijms21082888. Int J Mol Sci. 2020. PMID: 32326151 Free PMC article. Review.
-
The immunoregulatory role of alpha enolase in dendritic cell function during Chlamydia infection.BMC Immunol. 2017 May 19;18(1):27. doi: 10.1186/s12865-017-0212-1. BMC Immunol. 2017. PMID: 28525970 Free PMC article.
-
Hepatitis C Virus Alters Macrophage Cholesterol Metabolism Through Interaction with Scavenger Receptors.Viral Immunol. 2022 Apr;35(3):223-235. doi: 10.1089/vim.2021.0101. Viral Immunol. 2022. PMID: 35467430 Free PMC article.
-
Cortactin Interacts with Hepatitis C Virus Core and NS5A Proteins: Implications for Virion Assembly.J Virol. 2020 Sep 15;94(19):e01306-20. doi: 10.1128/JVI.01306-20. Print 2020 Sep 15. J Virol. 2020. PMID: 32727880 Free PMC article.
References
-
- Chen S, Yin P, Zhao X, Xing W, Hu C, Zhou L, et al. Serum lipid profiling of patients with chronic hepatitis B, cirrhosis, and hepatocellular carcinoma by ultra fast LC/IT-TOF MS. Electrophoresis. 2013;34(19):2848–56. Epub 2013/11/15. . - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
