

Translation readthrough mitigation
- PMID: 27281202
- PMCID: PMC5054982
- DOI: 10.1038/nature18308
Translation readthrough mitigation
Abstract
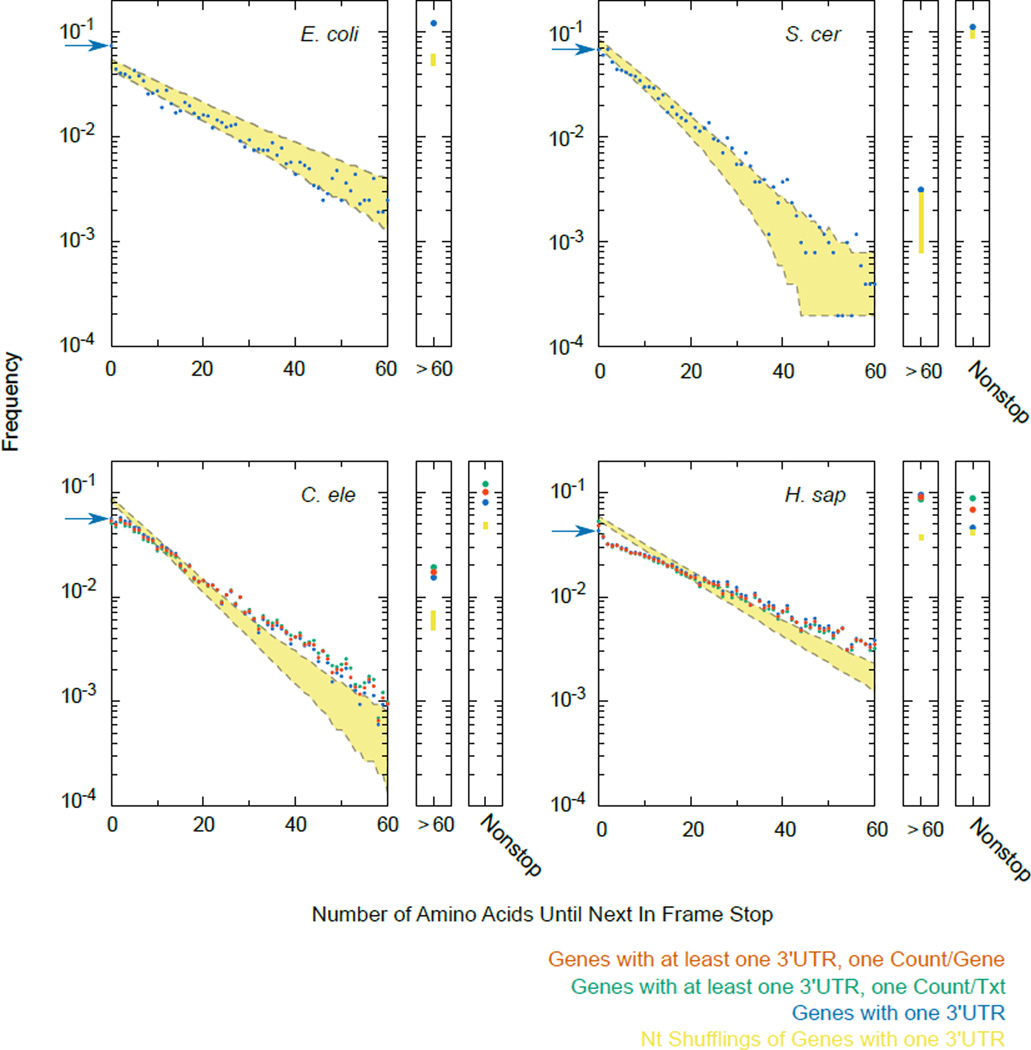
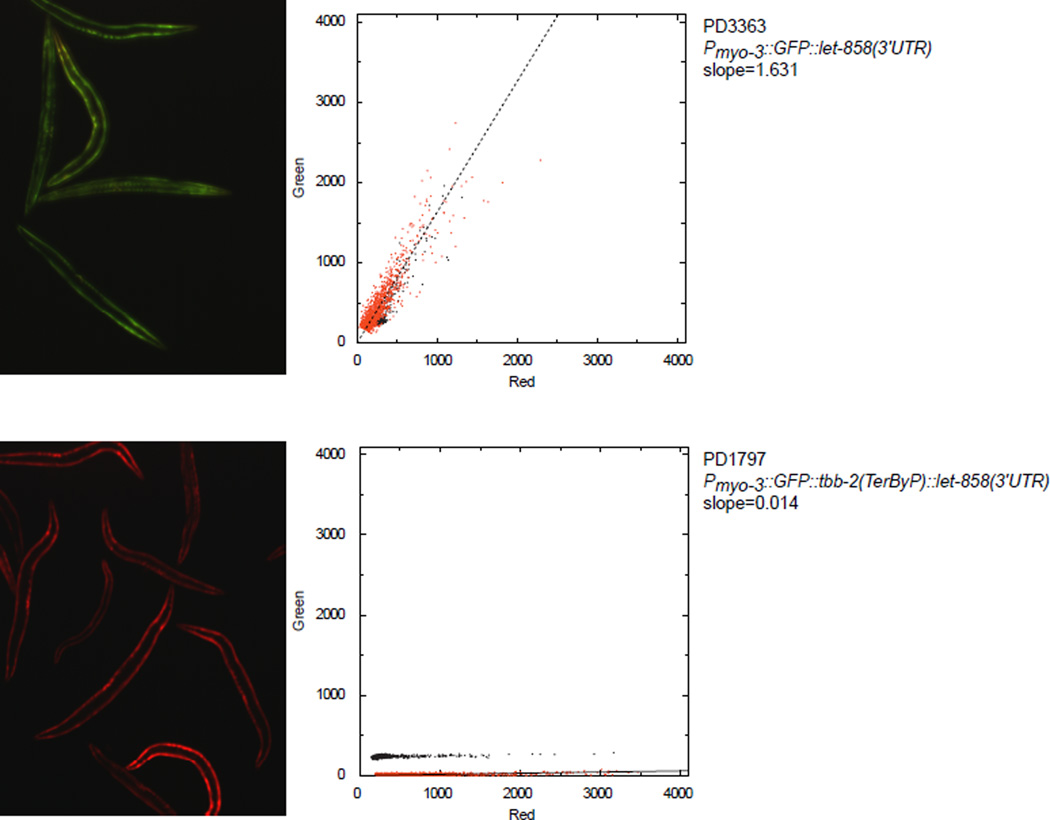
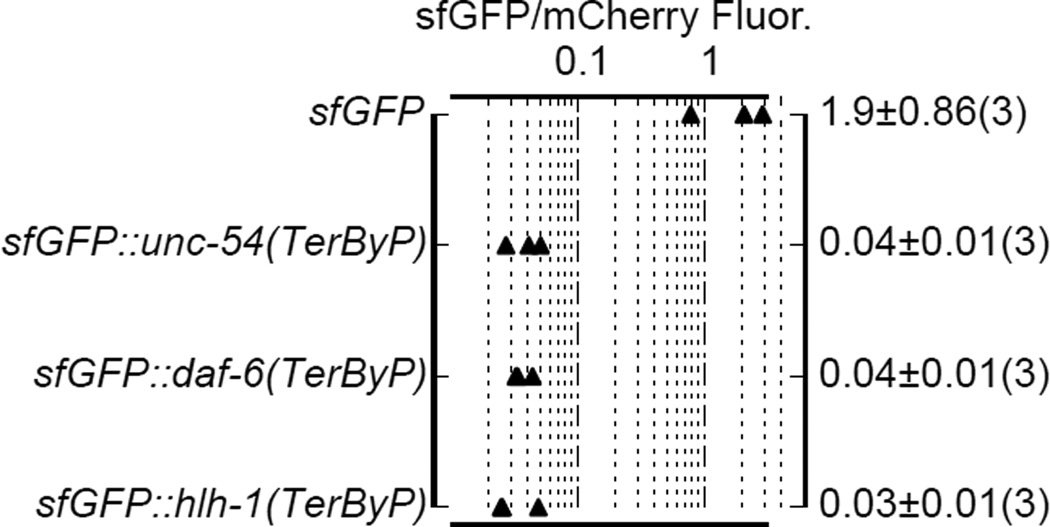
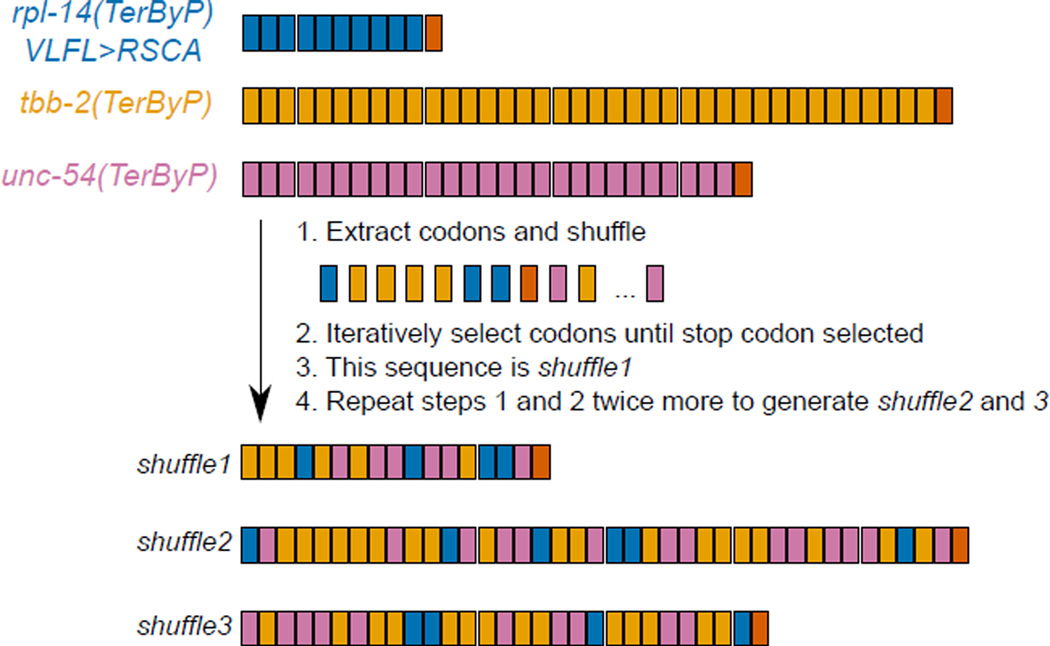
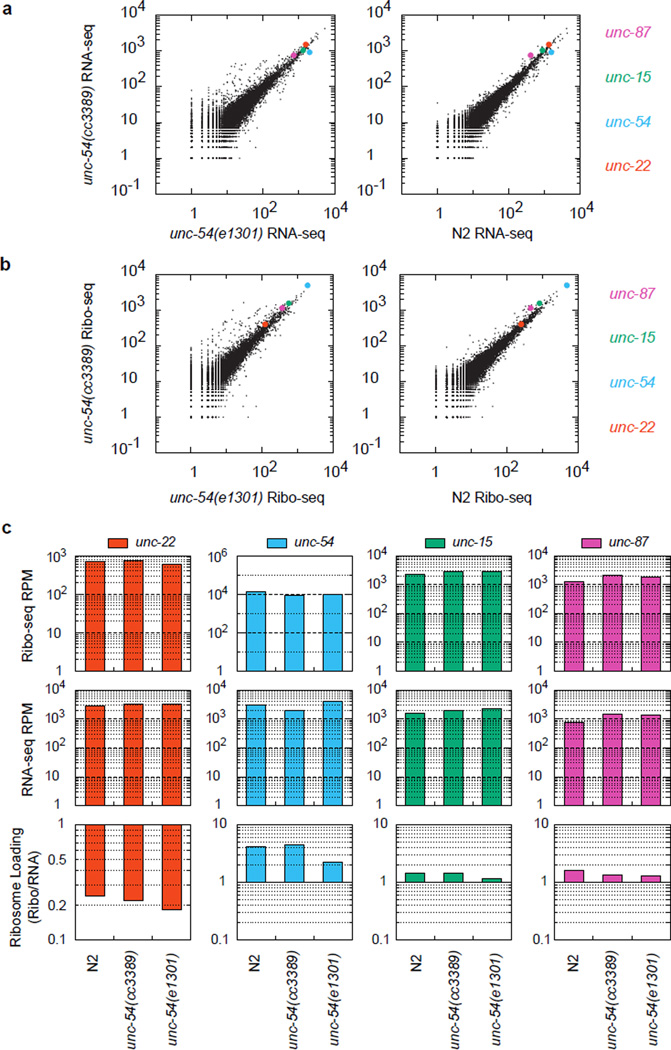
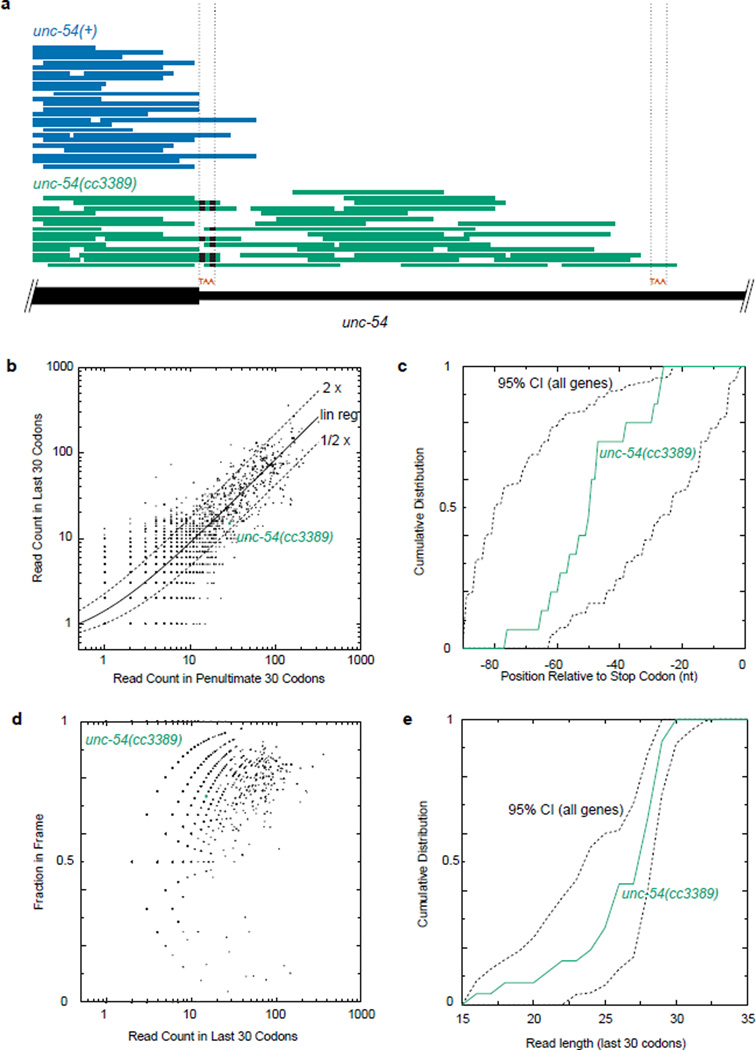
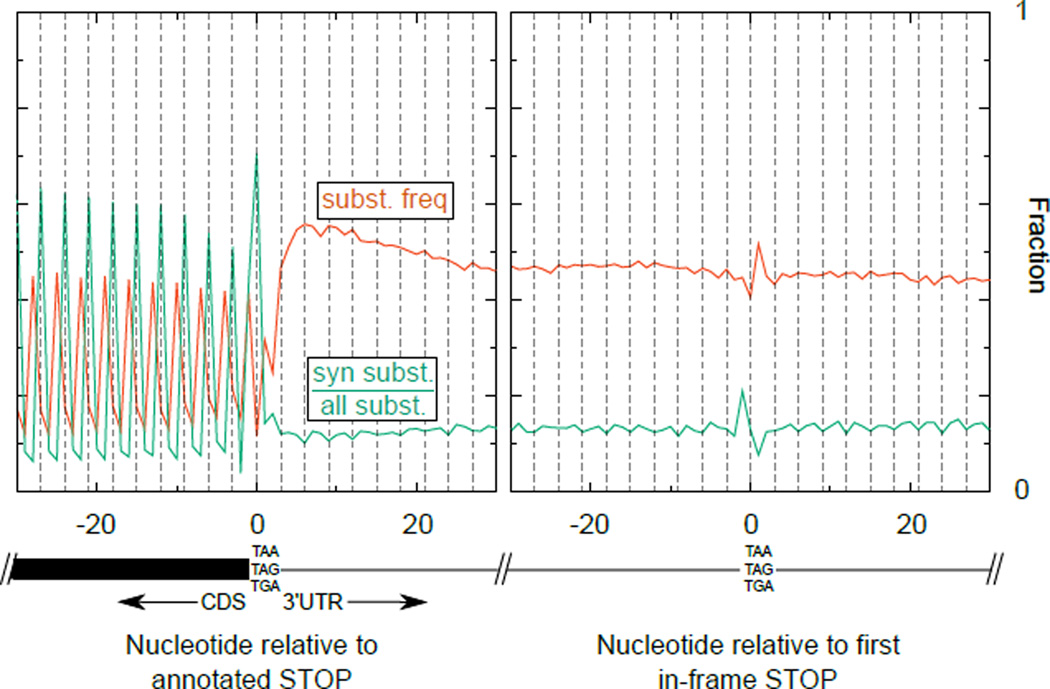
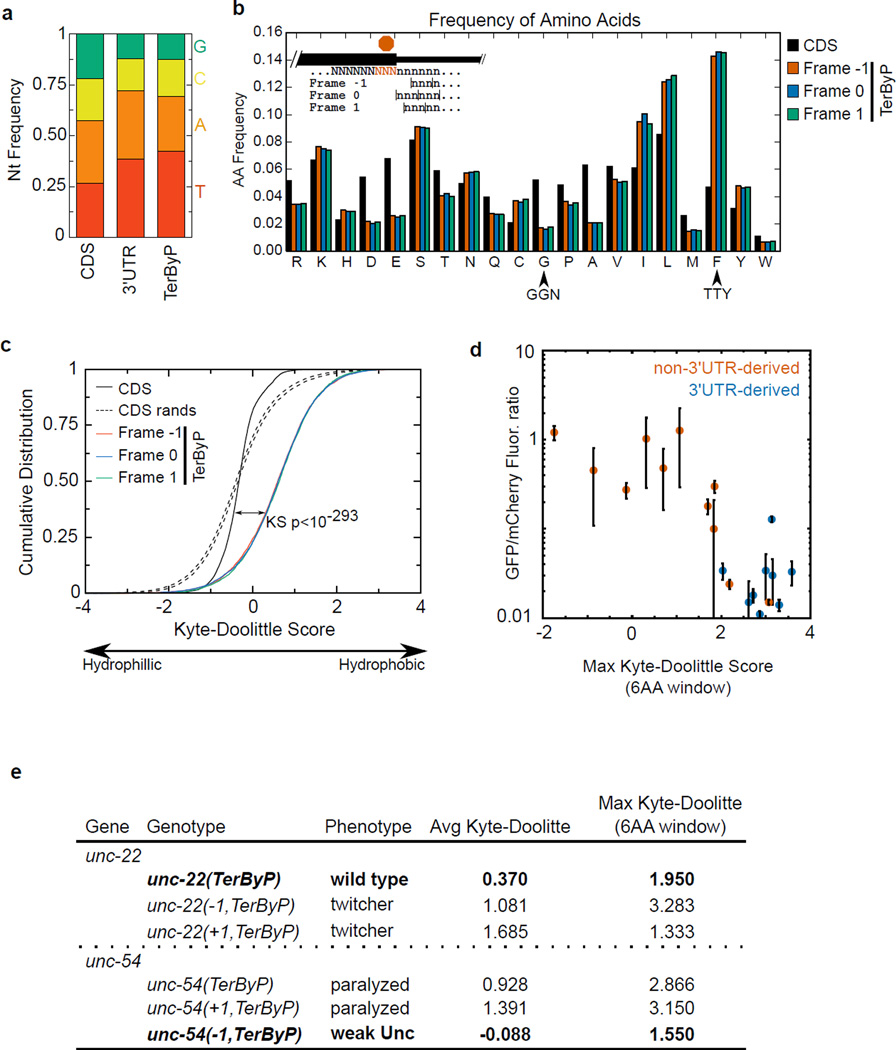
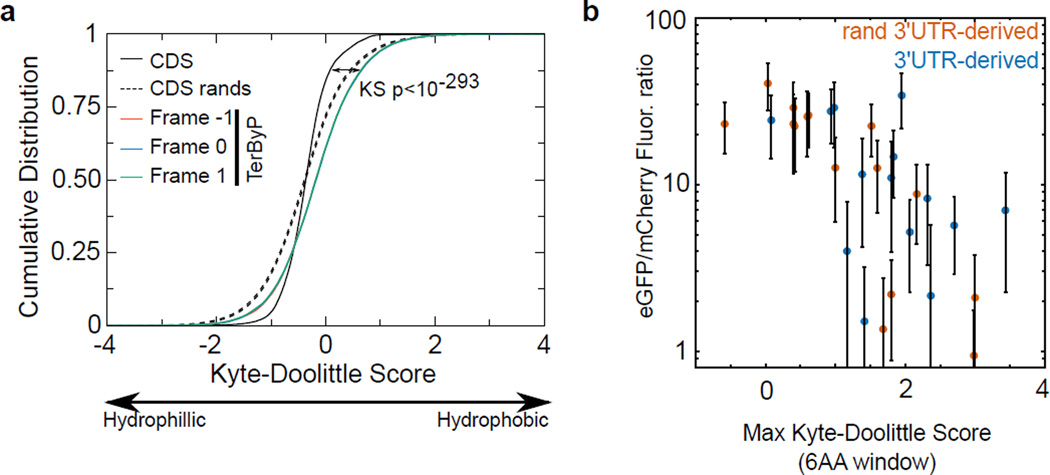
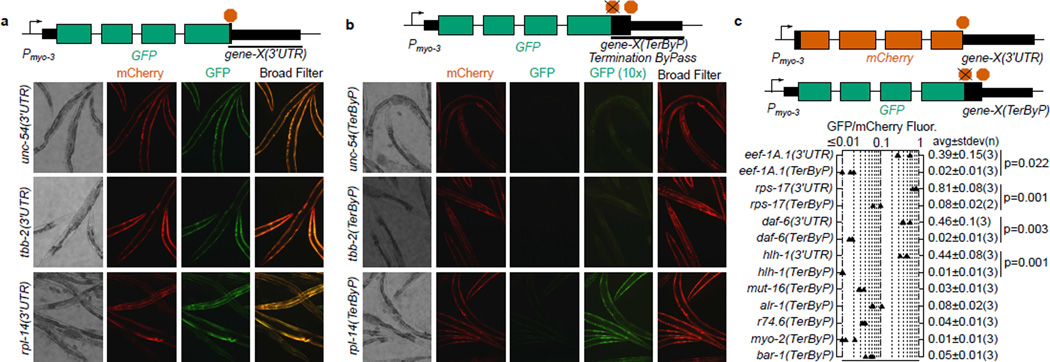
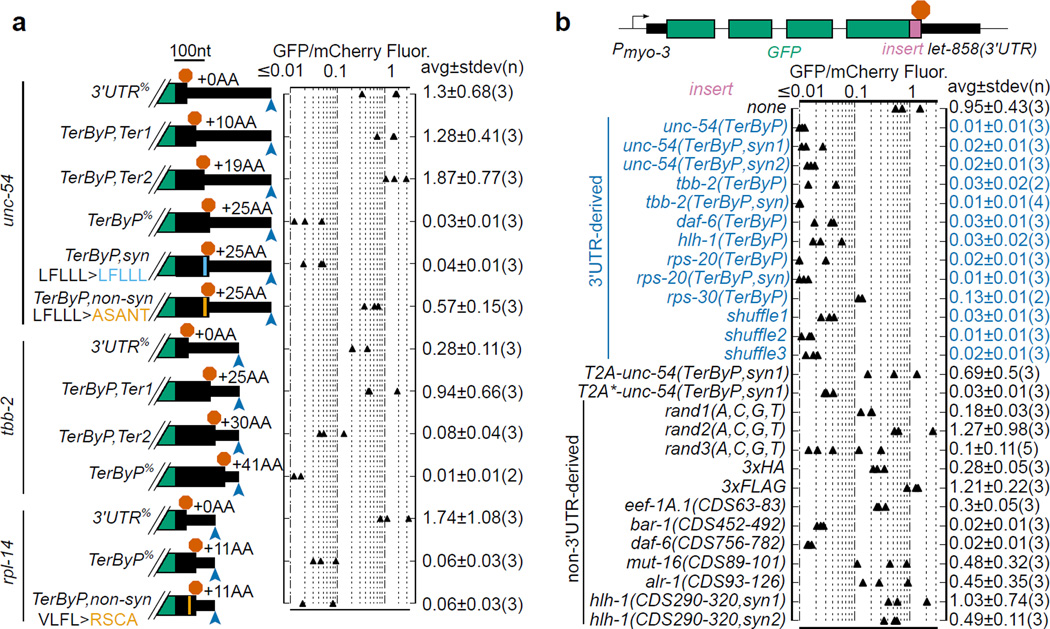
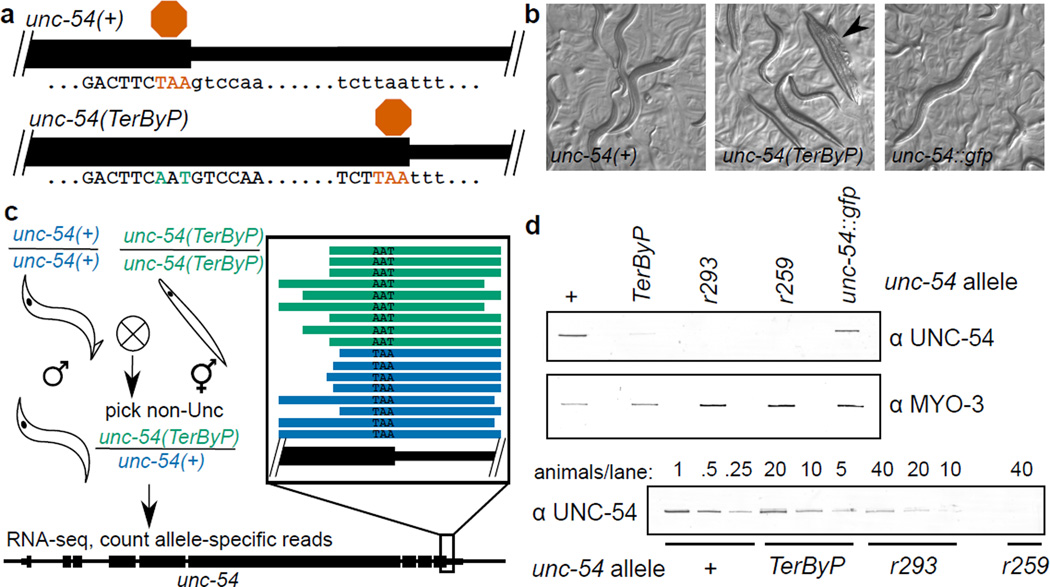
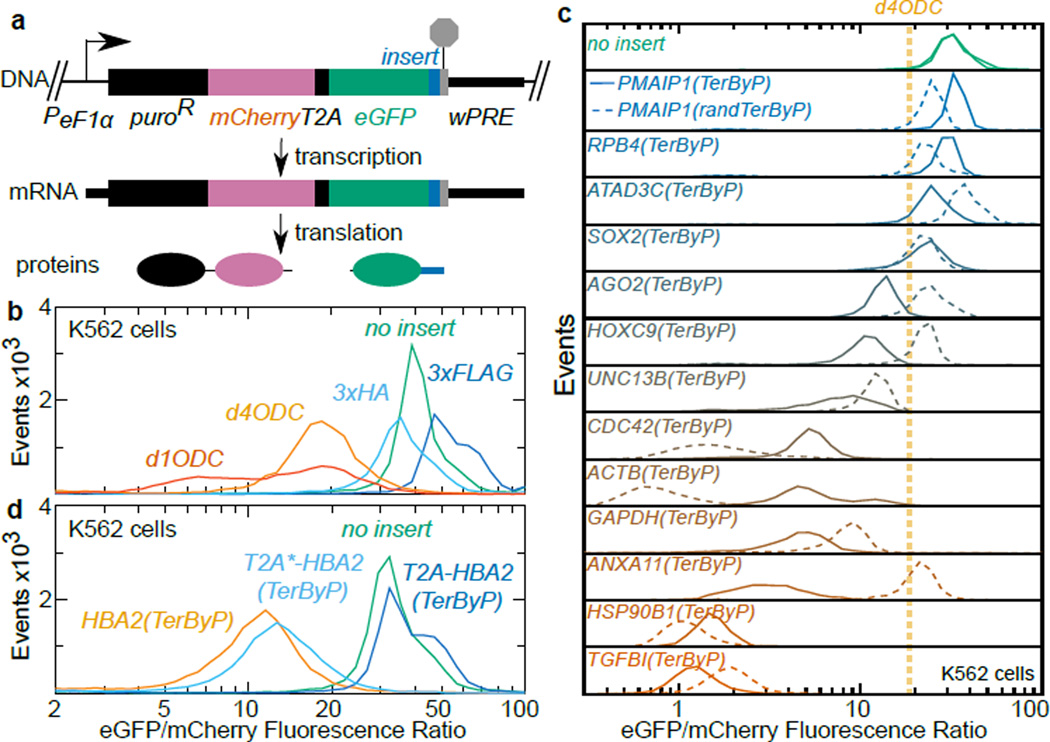
A fraction of ribosomes engaged in translation will fail to terminate when reaching a stop codon, yielding nascent proteins inappropriately extended on their C termini. Although such extended proteins can interfere with normal cellular processes, known mechanisms of translational surveillance are insufficient to protect cells from potential dominant consequences. Here, through a combination of transgenics and CRISPR–Cas9 gene editing in Caenorhabditis elegans, we demonstrate a consistent ability of cells to block accumulation of C-terminal-extended proteins that result from failure to terminate at stop codons. Sequences encoded by the 3′ untranslated region (UTR) were sufficient to lower protein levels. Measurements of mRNA levels and translation suggested a co- or post-translational mechanism of action for these sequences in C. elegans. Similar mechanisms evidently operate in human cells, in which we observed a comparable tendency for translated human 3′ UTR sequences to reduce mature protein expression in tissue culture assays, including 3′ UTR sequences from the hypomorphic ‘Constant Spring’ haemoglobin stop codon variant. We suggest that 3′ UTRs may encode peptide sequences that destabilize the attached protein, providing mitigation of unwelcome and varied translation errors.
Figures

References
-
- Falini B, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005;352:254–266. - PubMed
Extended References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
