

Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity
- PMID: 27286824
- PMCID: PMC4932939
- DOI: 10.1073/pnas.1523199113
Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity
Abstract
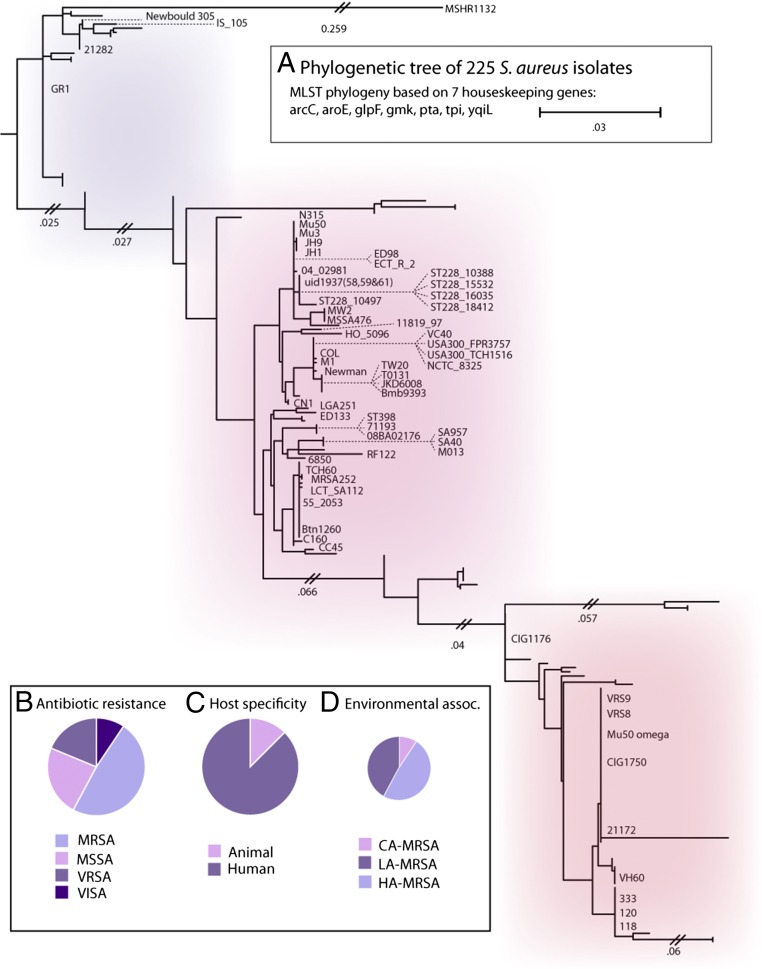
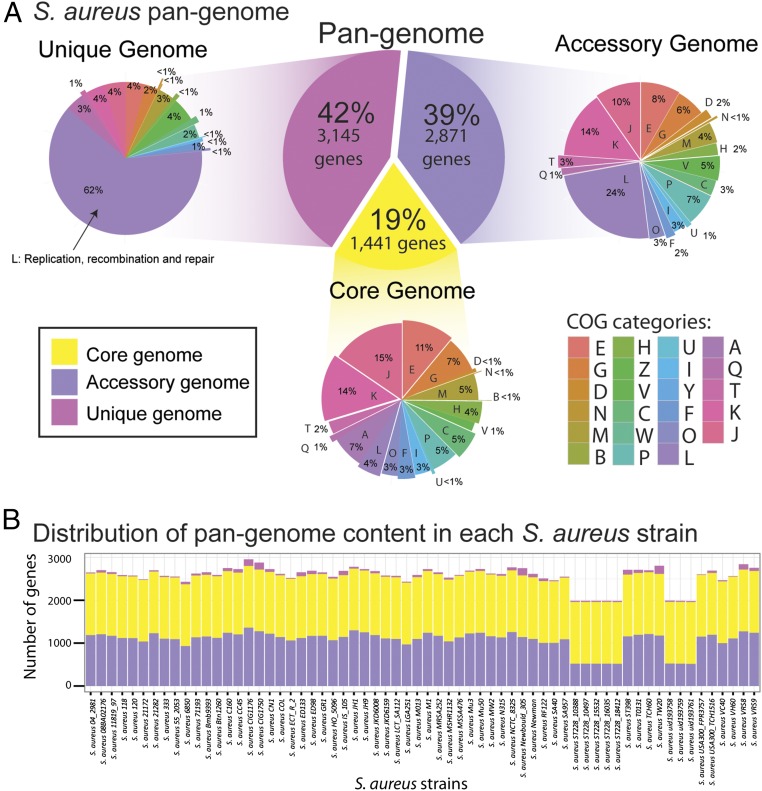
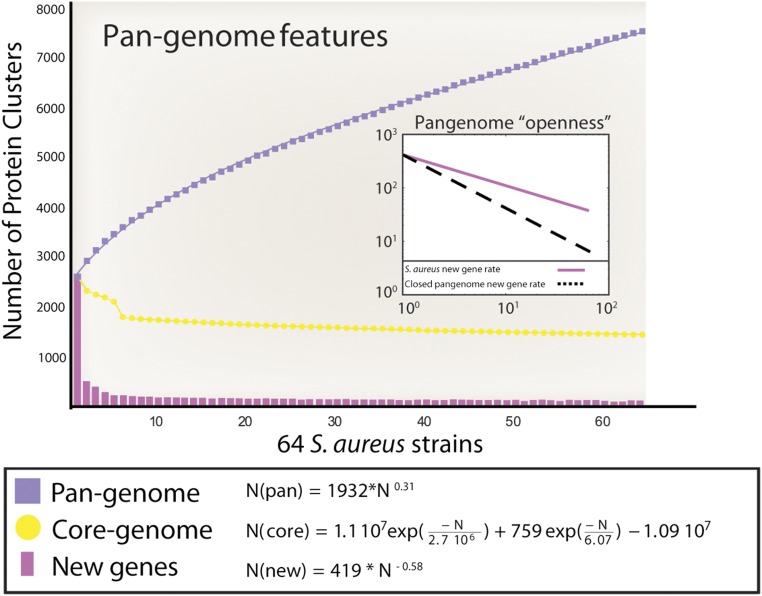
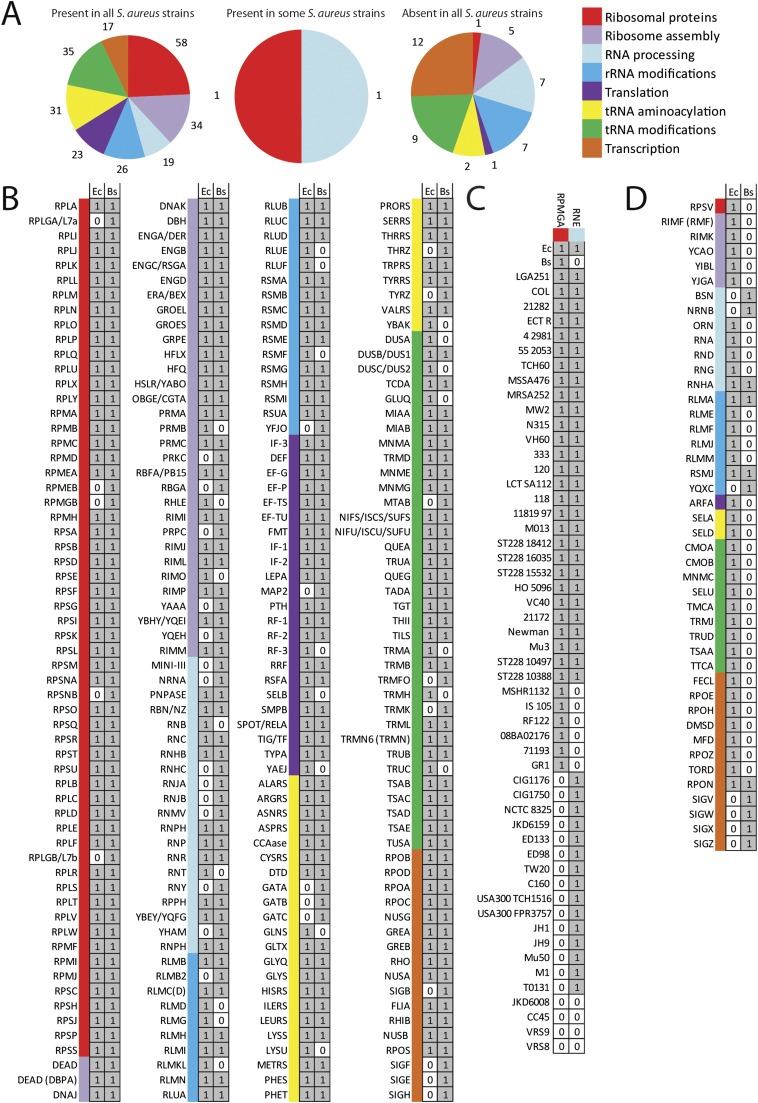
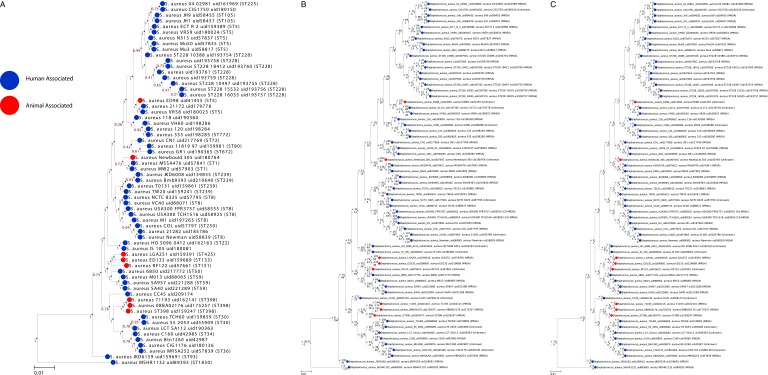
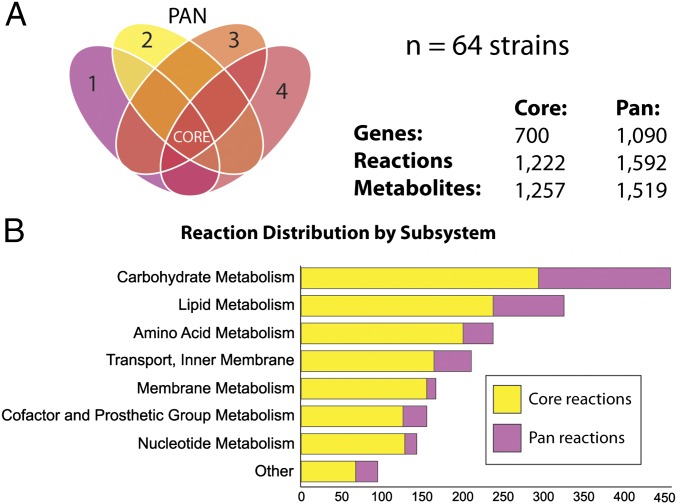
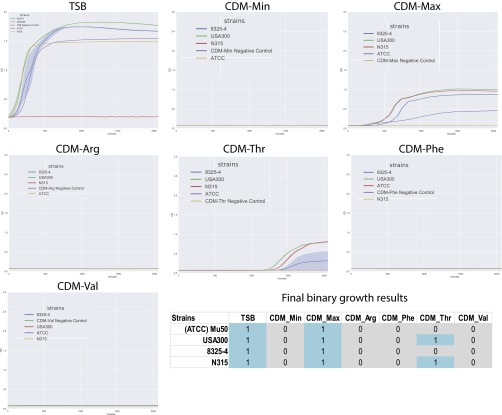
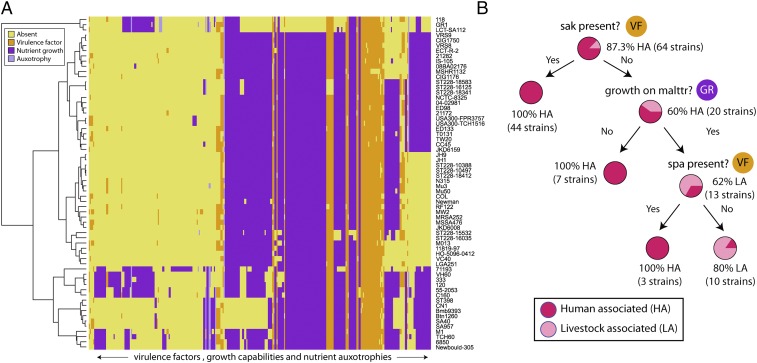
Staphylococcus aureus is a preeminent bacterial pathogen capable of colonizing diverse ecological niches within its human host. We describe here the pangenome of S. aureus based on analysis of genome sequences from 64 strains of S. aureus spanning a range of ecological niches, host types, and antibiotic resistance profiles. Based on this set, S. aureus is expected to have an open pangenome composed of 7,411 genes and a core genome composed of 1,441 genes. Metabolism was highly conserved in this core genome; however, differences were identified in amino acid and nucleotide biosynthesis pathways between the strains. Genome-scale models (GEMs) of metabolism were constructed for the 64 strains of S. aureus These GEMs enabled a systems approach to characterizing the core metabolic and panmetabolic capabilities of the S. aureus species. All models were predicted to be auxotrophic for the vitamins niacin (vitamin B3) and thiamin (vitamin B1), whereas strain-specific auxotrophies were predicted for riboflavin (vitamin B2), guanosine, leucine, methionine, and cysteine, among others. GEMs were used to systematically analyze growth capabilities in more than 300 different growth-supporting environments. The results identified metabolic capabilities linked to pathogenic traits and virulence acquisitions. Such traits can be used to differentiate strains responsible for mild vs. severe infections and preference for hosts (e.g., animals vs. humans). Genome-scale analysis of multiple strains of a species can thus be used to identify metabolic determinants of virulence and increase our understanding of why certain strains of this deadly pathogen have spread rapidly throughout the world.
Keywords: core genome; mathematical modeling; pangenome; pathogenicity; systems biology.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- van Belkum A, et al. Reclassification of Staphylococcus aureus nasal carriage types. J Infect Dis. 2009;199(12):1820–1826. - PubMed
-
- Hota B, et al. CDC Prevention Epicenters Predictors of clinical virulence in community-onset methicillin-resistant Staphylococcus aureus infections: The importance of USA300 and pneumonia. Clin Infect Dis. 2011;53(8):757–765. - PubMed
-
- Klevens RM, et al. Active Bacterial Core surveillance (ABCs) MRSA Investigators Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763–1771. - PubMed
-
- Otter JA, French GL. Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Europe. Lancet Infect Dis. 2010;10(4):227–239. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
