

Alport Syndrome in Women and Girls
- PMID: 27287265
- PMCID: PMC5012472
- DOI: 10.2215/CJN.00580116
Alport Syndrome in Women and Girls
Abstract
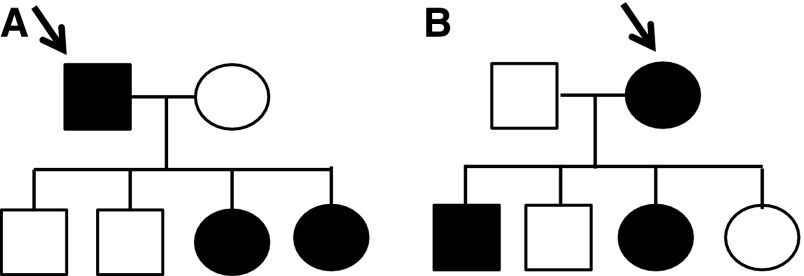
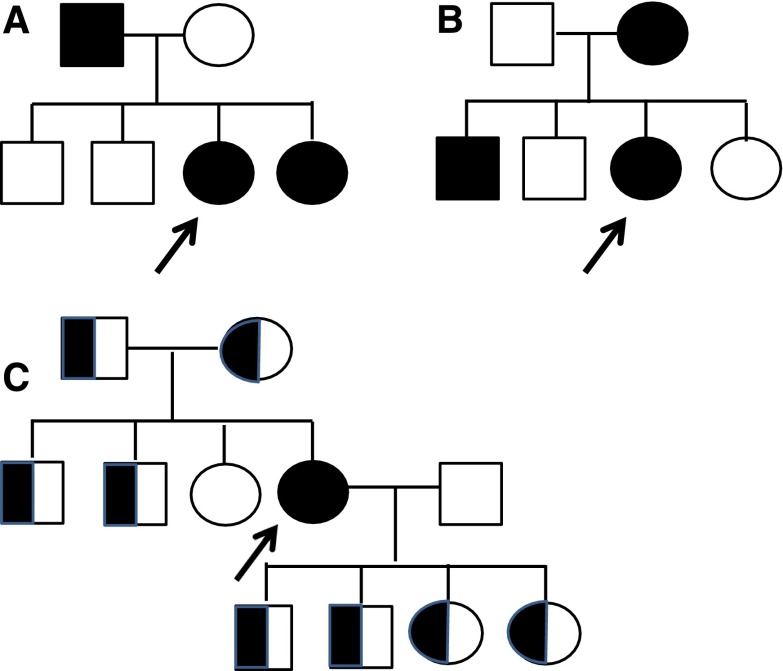
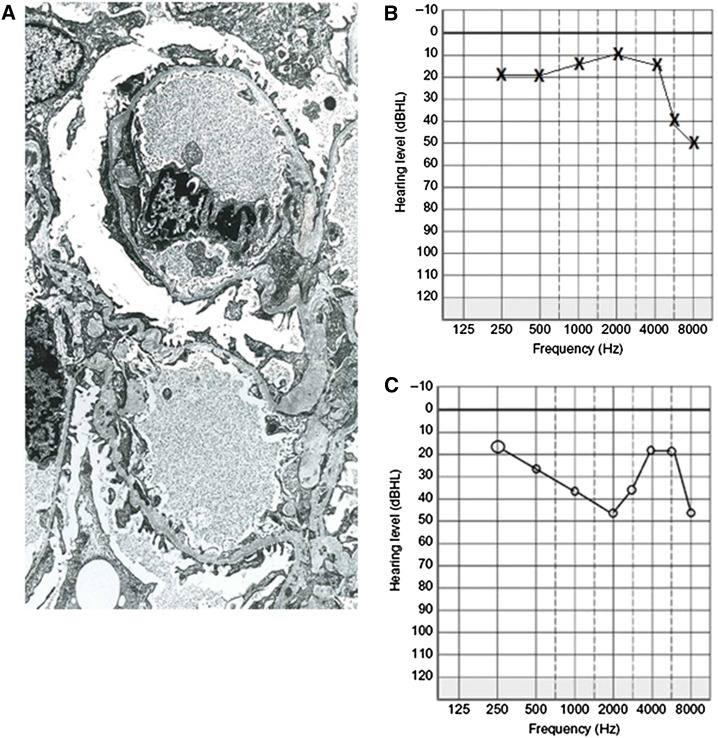
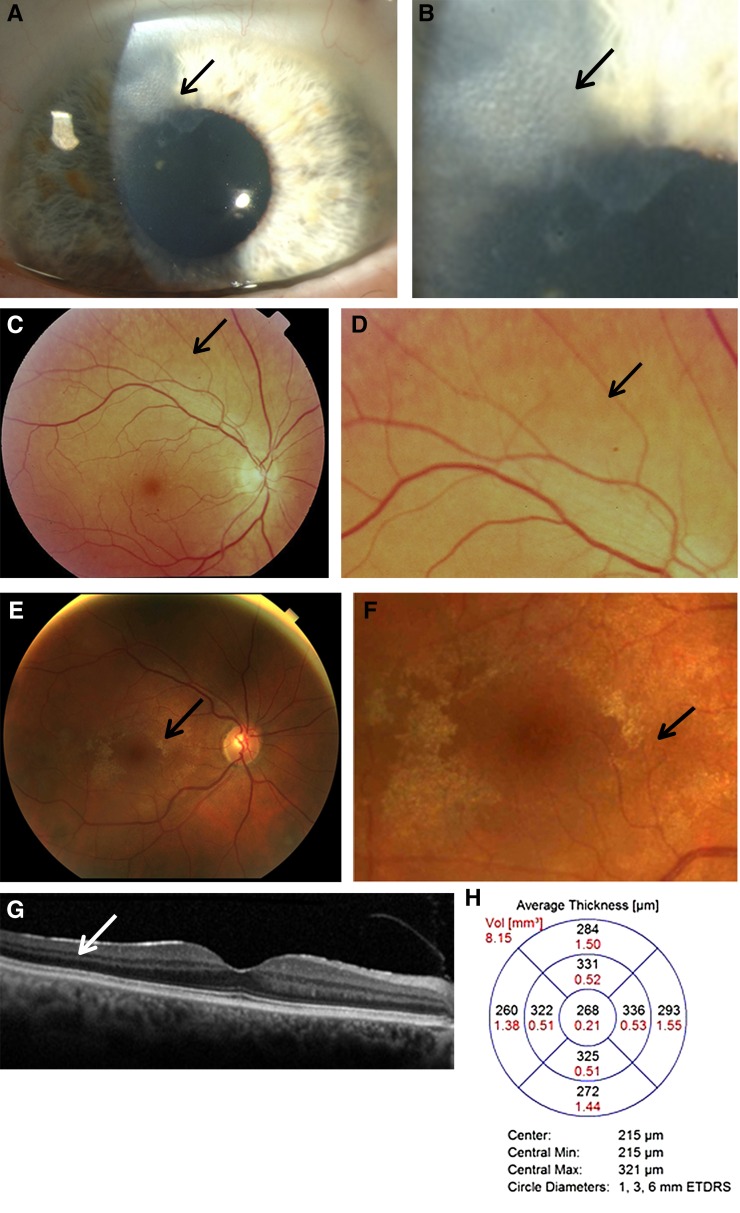
Alport syndrome is an inherited disease characterized by progressive renal failure, hearing loss, and ocular abnormalities. Inheritance is X-linked (85%) or autosomal recessive (15%). Many renal physicians think of Alport syndrome as primarily affecting men. However, twice as many women are affected by the X-linked diseases. Affected women are commonly undiagnosed, but 15%-30% develop renal failure by 60 years and often hearing loss by middle age. Half of their sons and daughters are also affected. Autosomal recessive Alport syndrome is less common, but is often mistaken for X-linked disease. Recessive inheritance is suspected where women develop early-onset renal failure or lenticonus. Their family may be consanguineous. The prognosis for other family members is very different from X-linked disease. Other generations, including parents and offspring, are not affected, and on average only one in four of their siblings inherit the disease. All women with Alport syndrome should have their diagnosis confirmed with genetic testing, even if their renal function is normal, because of their own risk of renal failure and the risk to their offspring. Their mutations indicate the mode of inheritance and the likelihood of disease transmission to their children, and the mutation type suggests the renal prognosis for both X-linked and recessive disease. Women with X-linked Alport syndrome should be tested at least annually for albuminuria and hypertension. The "Expert guidelines for the diagnosis and management of Alport syndrome" recommend treating those with albuminuria with renin-angiotensin-aldosterone system (RAAS) blockade (and adequate birth control because of the teratogenic risks of angiotensin converting enzyme inhibitors), believing that this will delay renal failure. Current recommendations are that women with autosomal recessive Alport syndrome should be treated with RAAS blockade from the time of diagnosis. In addition, women should be offered genetic counseling, informed of their reproductive options, and monitored closely during pregnancy for the development of hypertension.
Keywords: ACE inhibitors; Alport syndrome; Chronic; Deafness; Female; Genetic Testing; Hearing Loss; Hereditary; Humans; Kidney Failure; Mutation; Nephritis; albuminuria; genetic renal disease.
Copyright © 2016 by the American Society of Nephrology.
Figures
References
-
- Gubler M, Levy M, Broyer M, Naizot C, Gonzales G, Perrin D, Habib R: Alport’s syndrome. A report of 58 cases and a review of the literature. Am J Med 70: 493–505, 1981 - PubMed
-
- Habib R, Gubler MC, Hinglais N, Noël LH, Droz D, Levy M, Mahieu P, Foidart JM, Perrin D, Bois E, Grünfeld JP: Alport’s syndrome: experience at Hôpital Necker. Kidney Int Suppl 11: S20–S28, 1982 - PubMed
-
- Feingold J, Bois E, Chompret A, Broyer M, Gubler MC, Grünfeld JP: Genetic heterogeneity of Alport syndrome. Kidney Int 27: 672–677, 1985 - PubMed
-
- Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K: Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248: 1224–1227, 1990 - PubMed
-
- Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC, Pirson Y, Verellen-Dumoulin C, Chan B, Schröder CH, Smeets HJ, Reeder ST: Identification of mutations in the alpha 3(IV) and alpha 4(IV) collagen genes in autosomal recessive Alport syndrome. Nat Genet 8: 77–81, 1994 - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
