

Dynamic and Combinatorial Landscape of Histone Modifications during the Intraerythrocytic Developmental Cycle of the Malaria Parasite
- PMID: 27291344
- PMCID: PMC5905347
- DOI: 10.1021/acs.jproteome.6b00366
Dynamic and Combinatorial Landscape of Histone Modifications during the Intraerythrocytic Developmental Cycle of the Malaria Parasite
Abstract
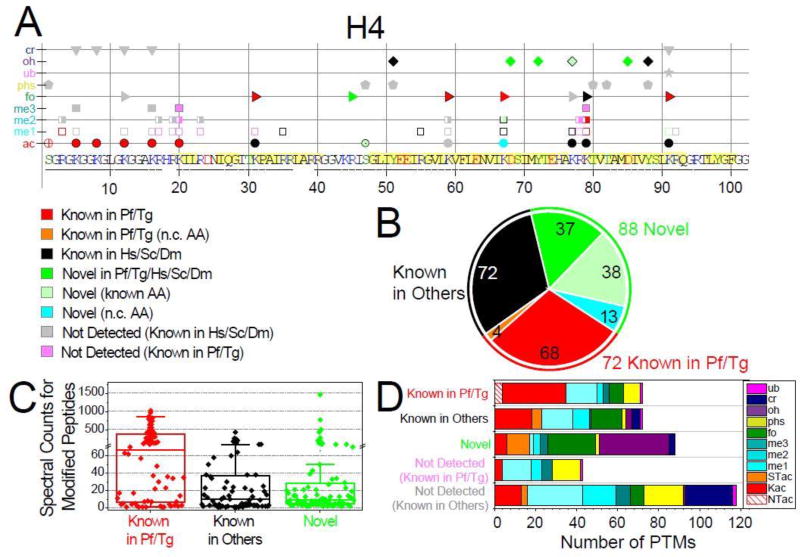
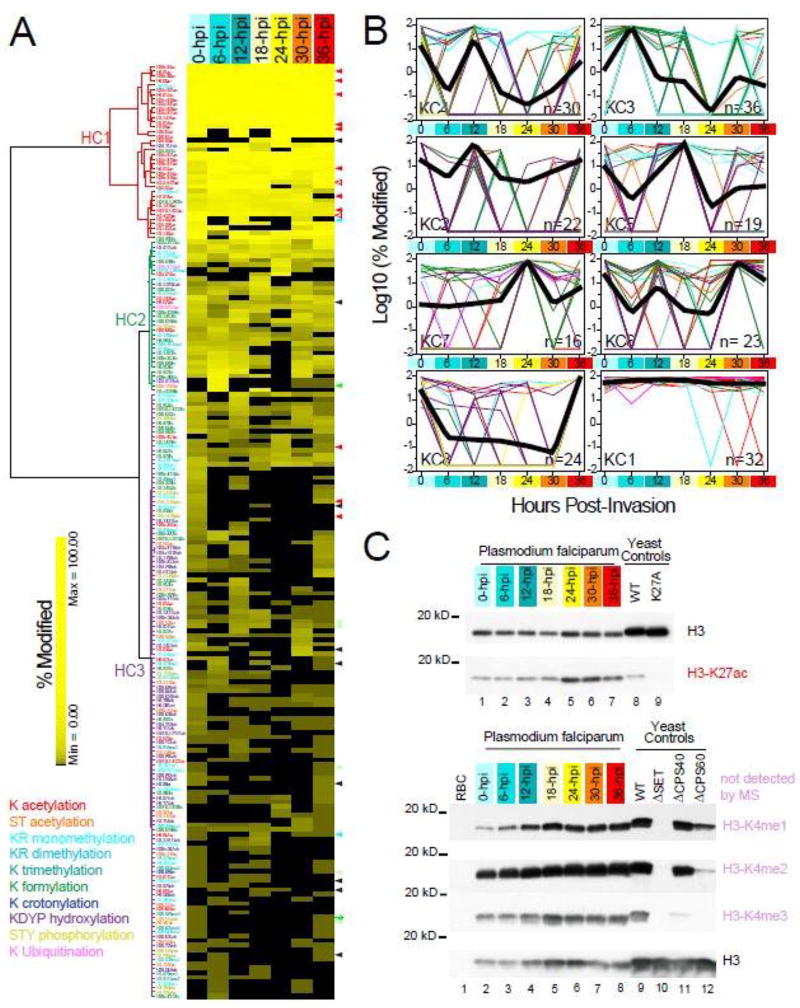
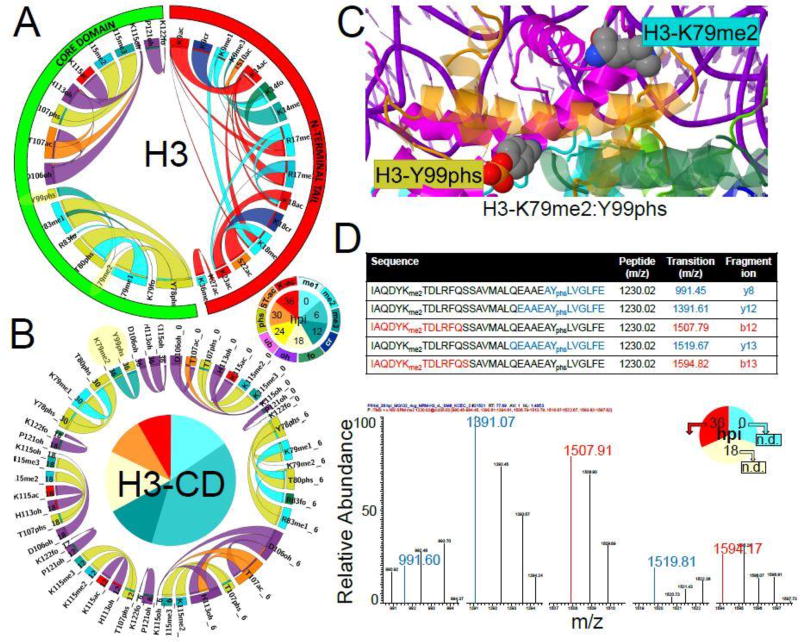
A major obstacle in understanding the complex biology of the malaria parasite remains to discover how gene transcription is controlled during its life cycle. Accumulating evidence indicates that the parasite's epigenetic state plays a fundamental role in gene expression and virulence. Using a comprehensive and quantitative mass spectrometry approach, we determined the global and dynamic abundance of histones and their covalent post-transcriptional modifications throughout the intraerythrocytic developmental cycle of Plasmodium falciparum. We detected a total of 232 distinct modifications, of which 160 had never been detected in Plasmodium and 88 had never been identified in any other species. We further validated over 10% of the detected modifications and their expression patterns by multiple reaction monitoring assays. In addition, we uncovered an unusual chromatin organization with parasite-specific histone modifications and combinatorial dynamics that may be directly related to transcriptional activity, DNA replication, and cell cycle progression. Overall, our data suggest that the malaria parasite has a unique histone modification signature that correlates with parasite virulence.
Keywords: cell cycle; epigenetics; histones; label-free quantification; malaria; multiple reaction monitoring; parasite; post-translational modifications; tandem mass spectrometry.
Figures

References
-
- Martin C, Zhang Y. Mechanisms of epigenetic inheritance. Curr Opin Cell Biol. 2007;19(3):266–72. - PubMed
-
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–28. - PMC - PubMed
-
- Rivera C, Gurard-Levin ZA, Almouzni G, Loyola A. Histone lysine methylation and chromatin replication. Biochim Biophys Acta. 2014;1839(12):1433–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
