

MTHFD1 controls DNA methylation in Arabidopsis
- PMID: 27291711
- PMCID: PMC4909953
- DOI: 10.1038/ncomms11640
MTHFD1 controls DNA methylation in Arabidopsis
Abstract
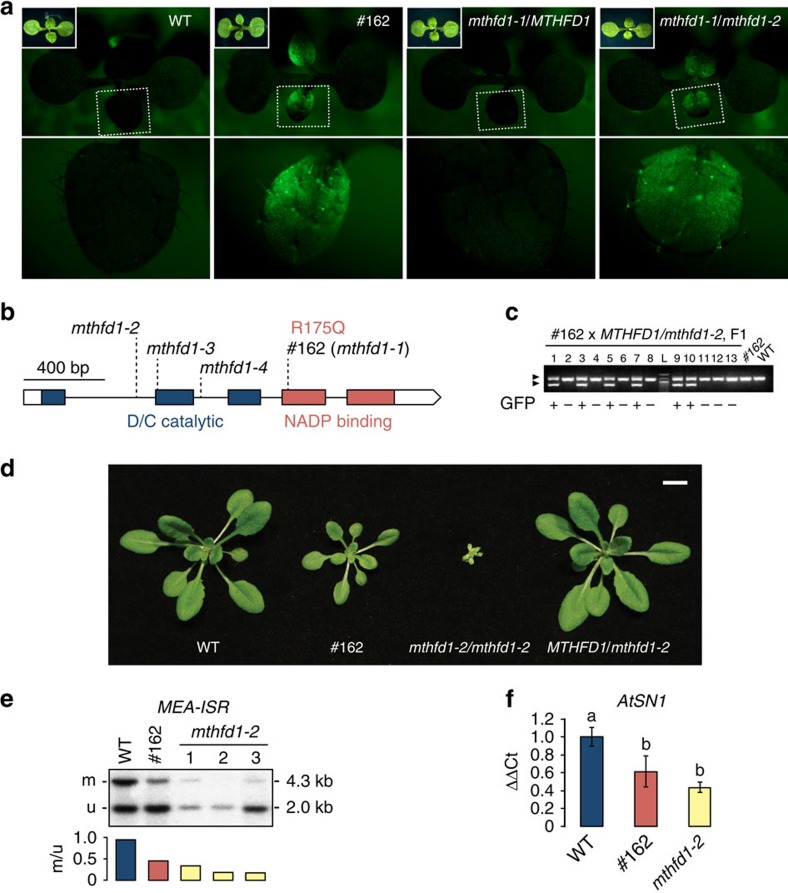
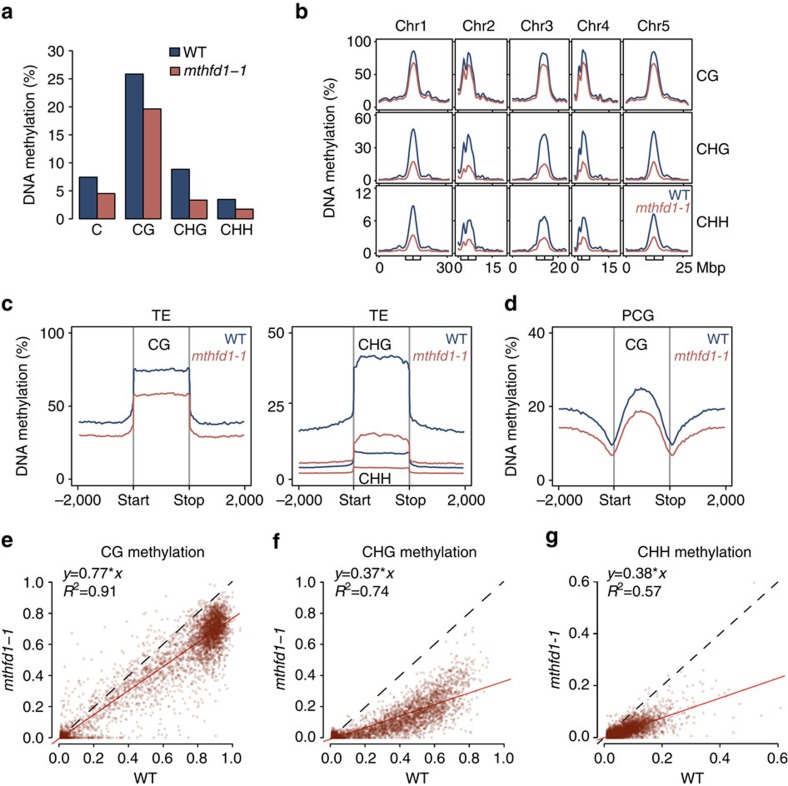
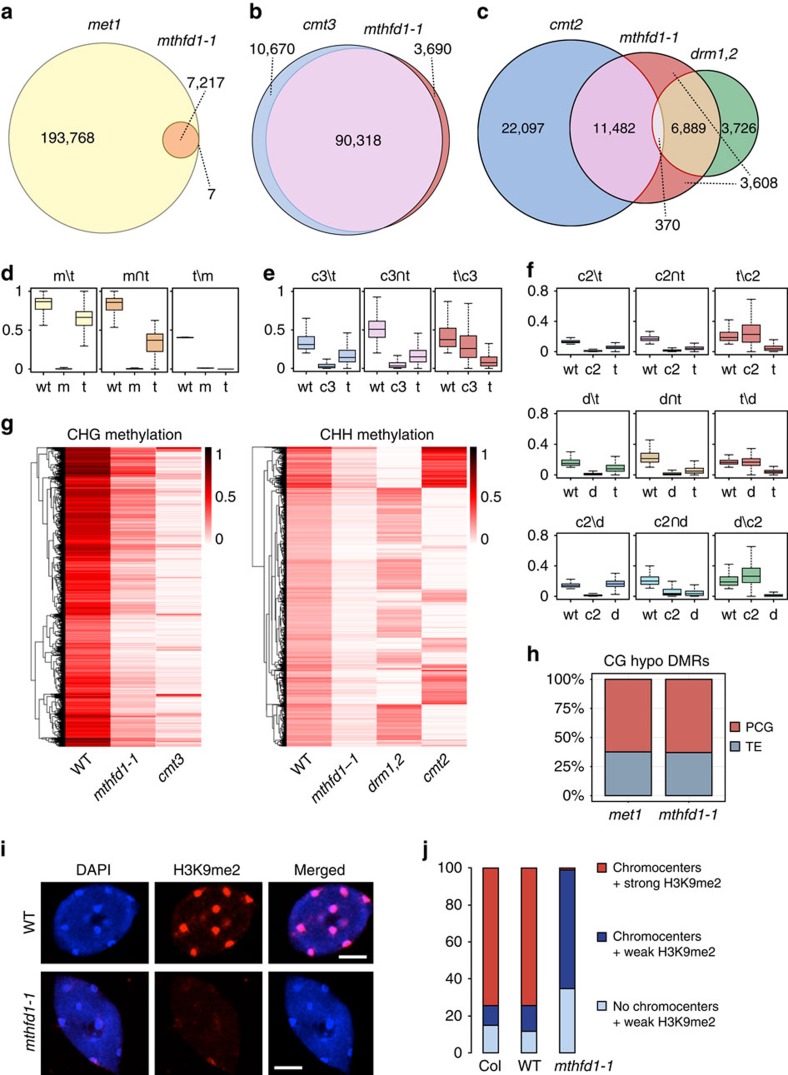
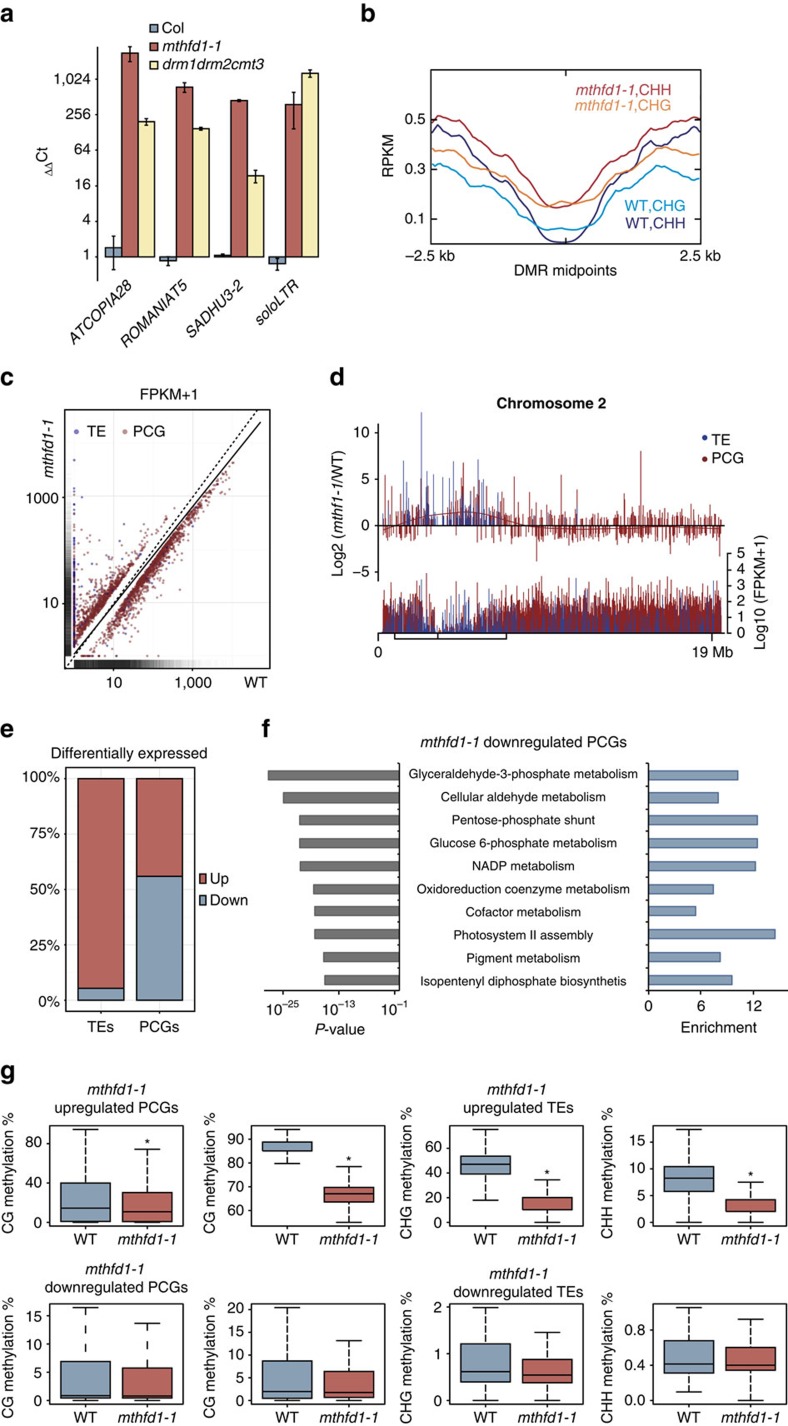
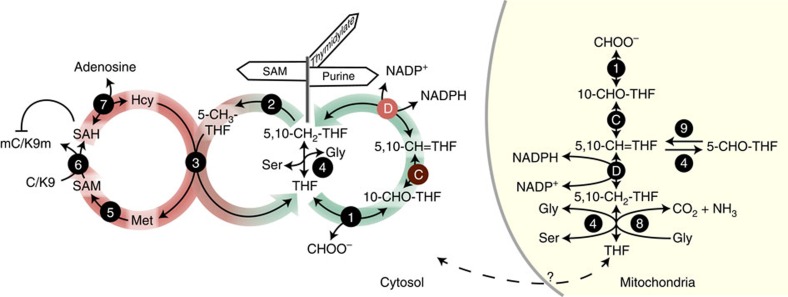
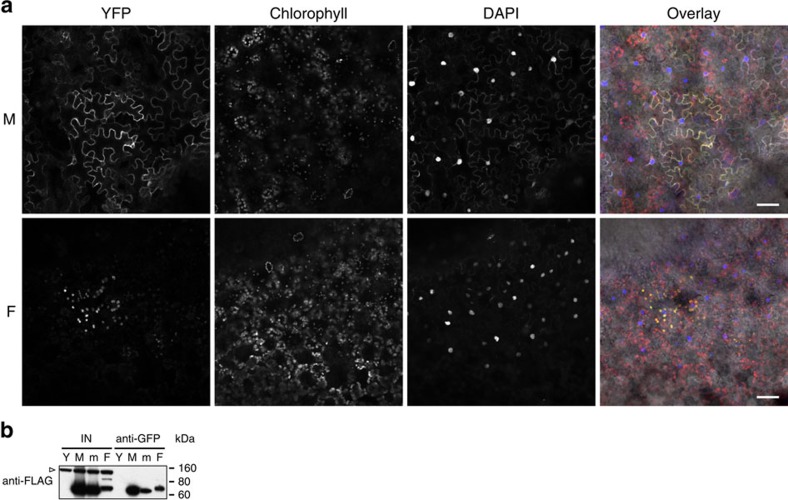
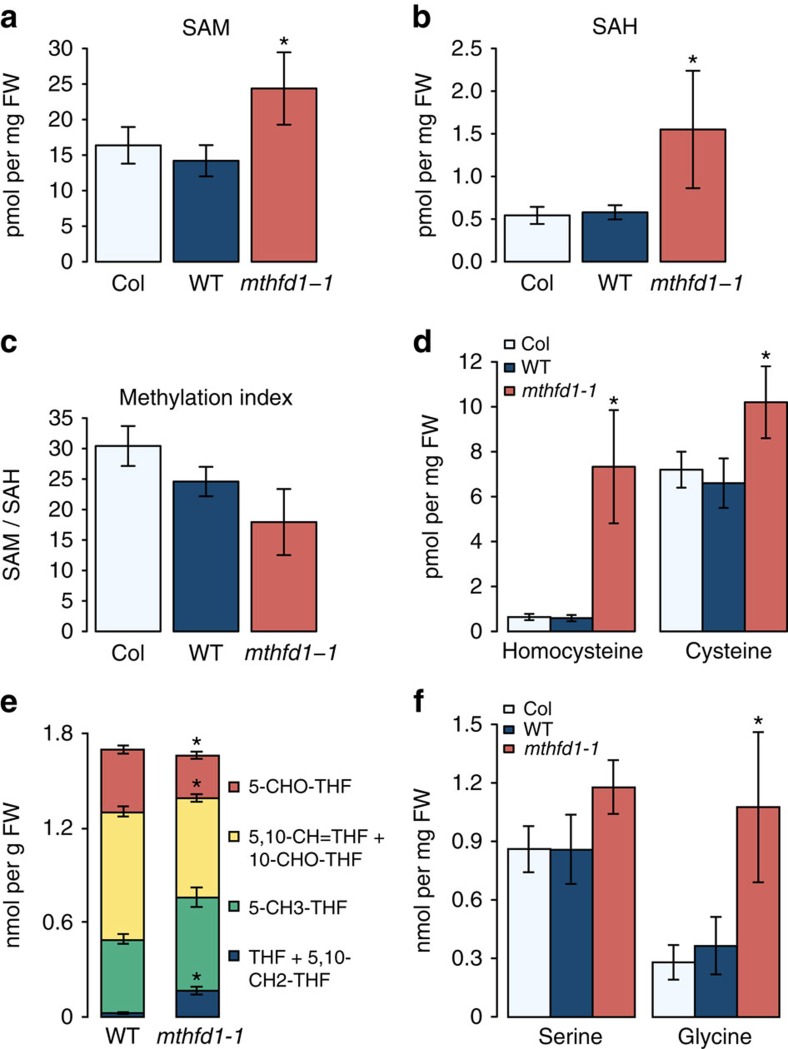
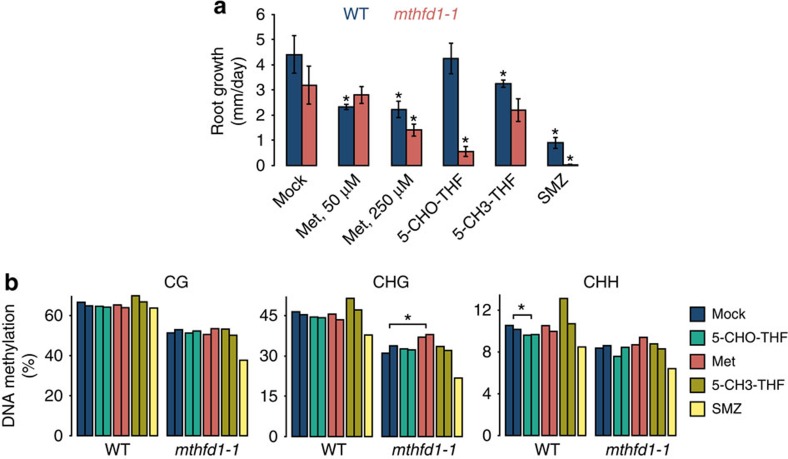
DNA methylation is an epigenetic mechanism that has important functions in transcriptional silencing and is associated with repressive histone methylation (H3K9me). To further investigate silencing mechanisms, we screened a mutagenized Arabidopsis thaliana population for expression of SDCpro-GFP, redundantly controlled by DNA methyltransferases DRM2 and CMT3. Here, we identify the hypomorphic mutant mthfd1-1, carrying a mutation (R175Q) in the cytoplasmic bifunctional methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase (MTHFD1). Decreased levels of oxidized tetrahydrofolates in mthfd1-1 and lethality of loss-of-function demonstrate the essential enzymatic role of MTHFD1 in Arabidopsis. Accumulation of homocysteine and S-adenosylhomocysteine, genome-wide DNA hypomethylation, loss of H3K9me and transposon derepression indicate that S-adenosylmethionine-dependent transmethylation is inhibited in mthfd1-1. Comparative analysis of DNA methylation revealed that the CMT3 and CMT2 pathways involving positive feedback with H3K9me are mostly affected. Our work highlights the sensitivity of epigenetic networks to one-carbon metabolism due to their common S-adenosylmethionine-dependent transmethylation and has implications for human MTHFD1-associated diseases.
Figures
References
-
- Cao X. & Jacobsen S. E. Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12, 1138–1144 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
