

Protein-structure-guided discovery of functional mutations across 19 cancer types
- PMID: 27294619
- PMCID: PMC5315576
- DOI: 10.1038/ng.3586
Protein-structure-guided discovery of functional mutations across 19 cancer types
Erratum in
-
Corrigendum: Protein-structure-guided discovery of functional mutations across 19 cancer types.Nat Genet. 2017 Jul 27;49(8):1286. doi: 10.1038/ng0817-1286a. Nat Genet. 2017. PMID: 28747753 No abstract available.
Abstract
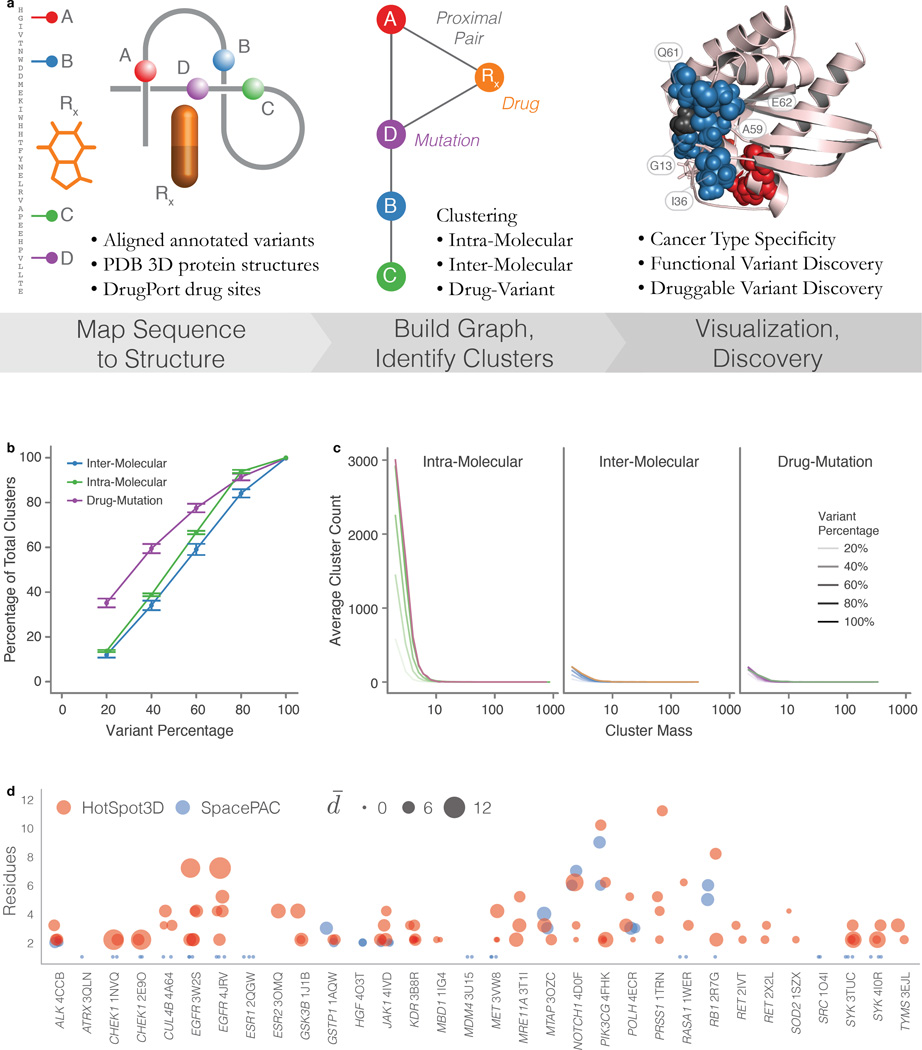
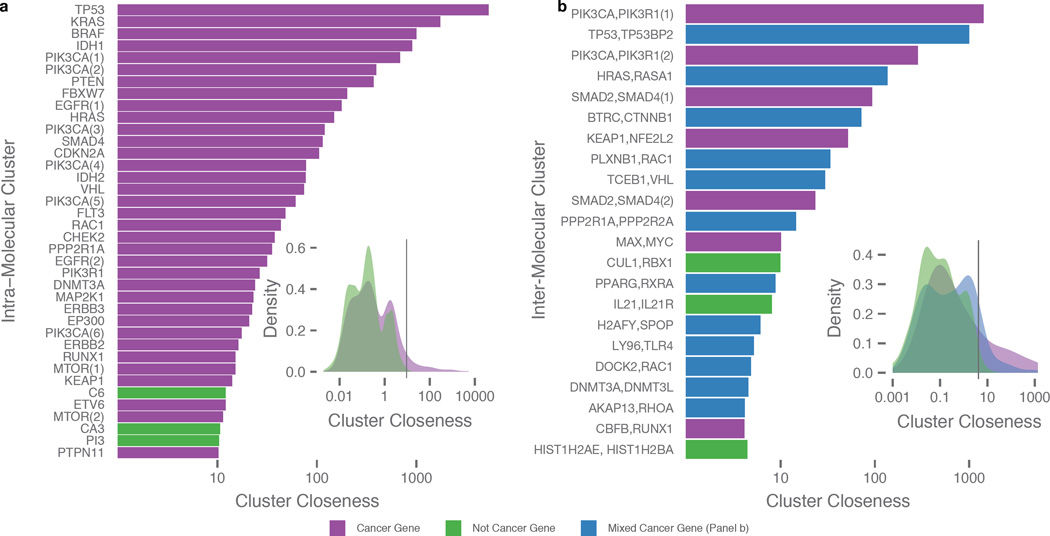
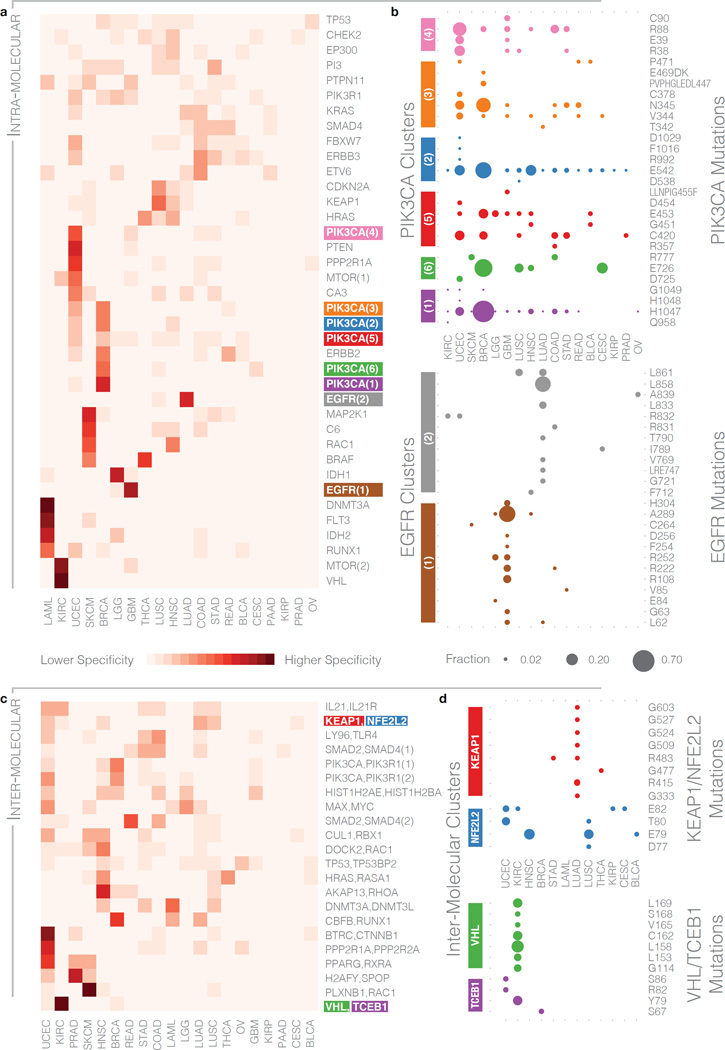
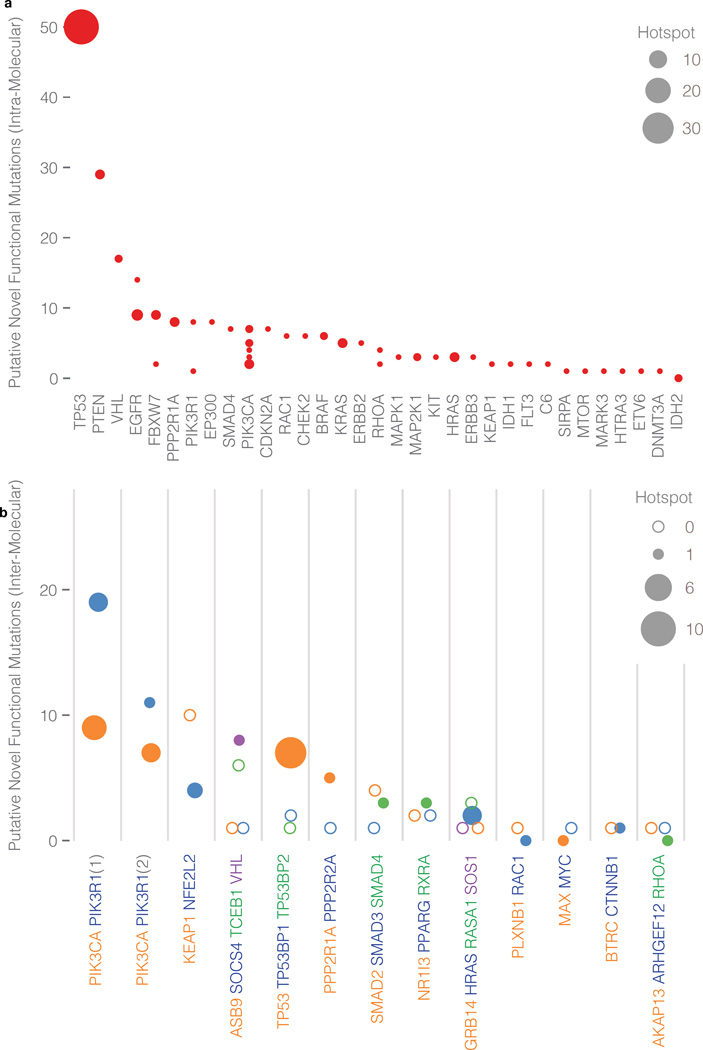
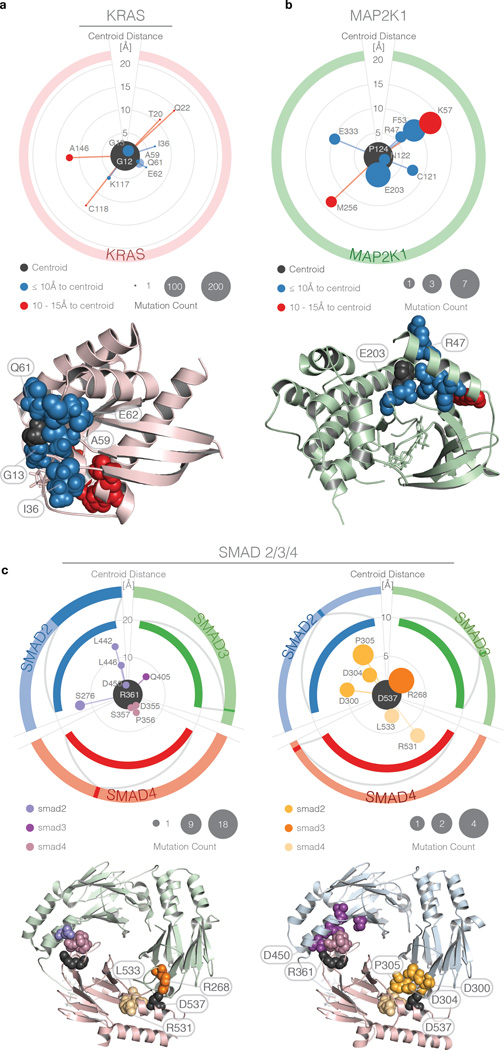
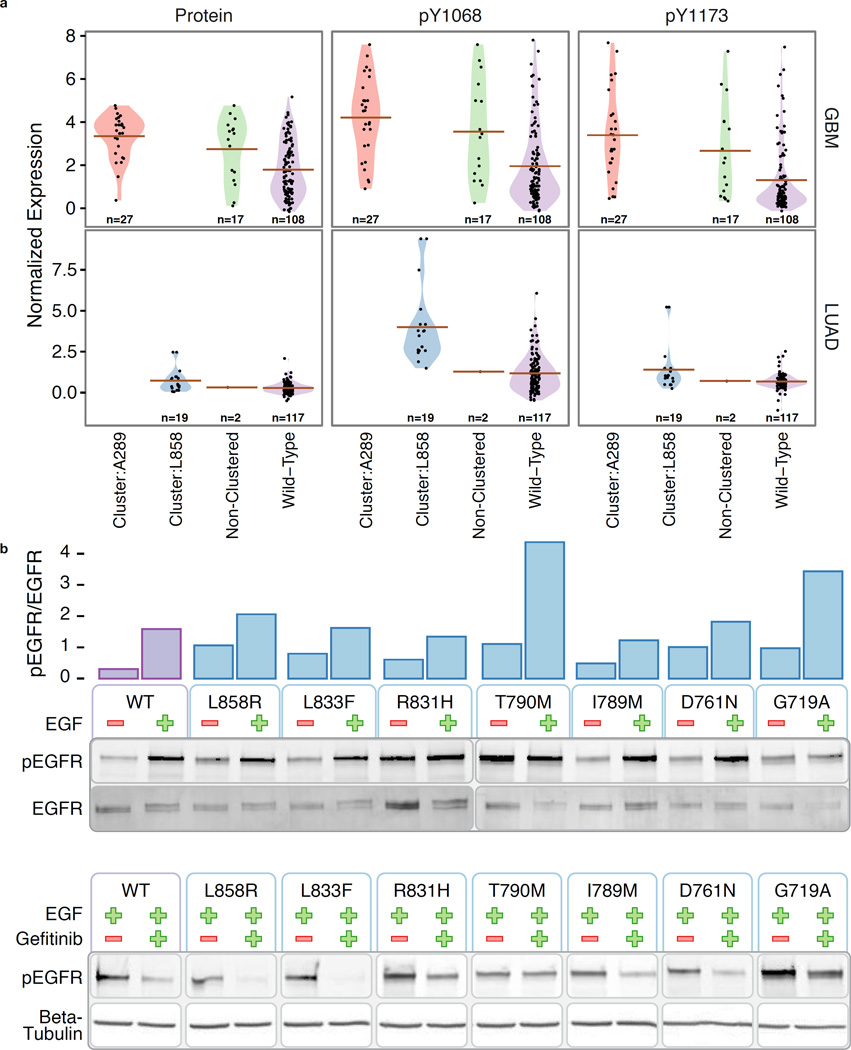
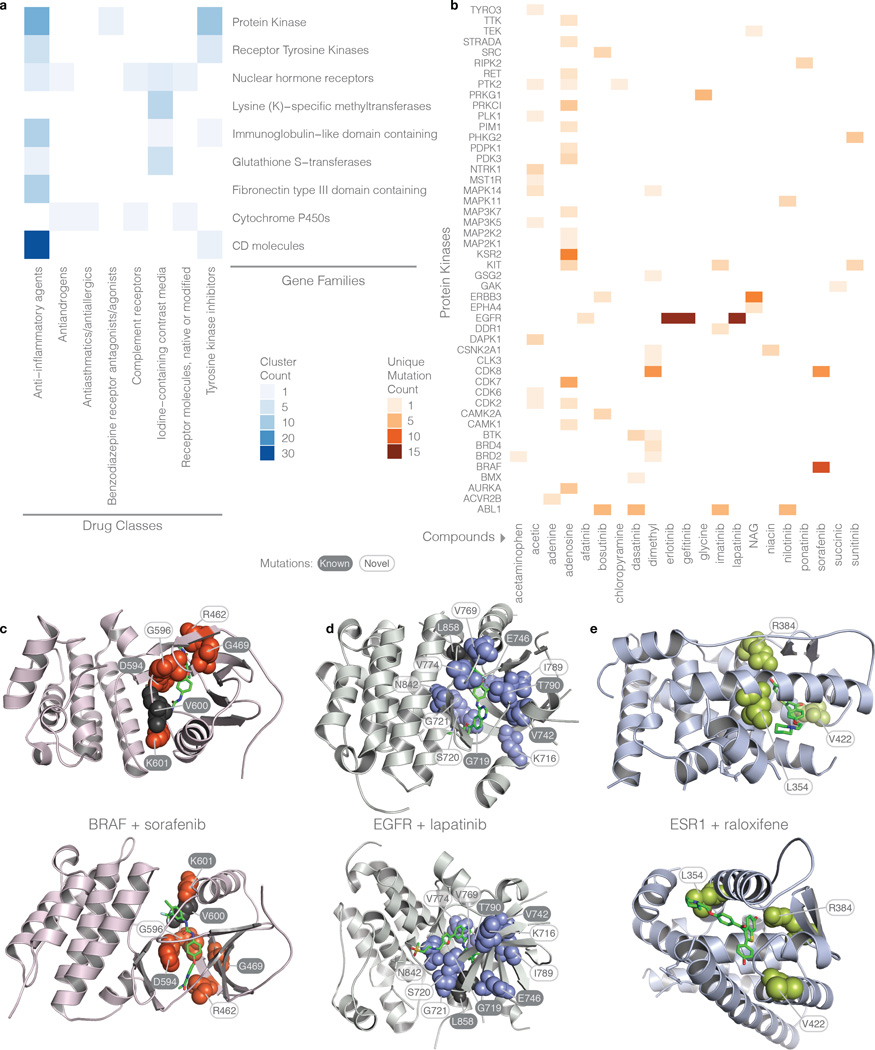
Local concentrations of mutations are well known in human cancers. However, their three-dimensional spatial relationships in the encoded protein have yet to be systematically explored. We developed a computational tool, HotSpot3D, to identify such spatial hotspots (clusters) and to interpret the potential function of variants within them. We applied HotSpot3D to >4,400 TCGA tumors across 19 cancer types, discovering >6,000 intra- and intermolecular clusters, some of which showed tumor and/or tissue specificity. In addition, we identified 369 rare mutations in genes including TP53, PTEN, VHL, EGFR, and FBXW7 and 99 medium-recurrence mutations in genes such as RUNX1, MTOR, CA3, PI3, and PTPN11, all mapping within clusters having potential functional implications. As a proof of concept, we validated our predictions in EGFR using high-throughput phosphorylation data and cell-line-based experimental evaluation. Finally, mutation-drug cluster and network analysis predicted over 800 promising candidates for druggable mutations, raising new possibilities for designing personalized treatments for patients carrying specific mutations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Methods References
-
- Berman HM. The Protein Data Bank: a historical perspective. Acta crystallographica. Section A, Foundations of crystallography. 2008;64:88–95. - PubMed
-
- Dangalchev C. Residual closeness in networks. Physica A: Statistical Mechanics and its Applications. 2006;365:556–564.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
