

Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling
- PMID: 27295076
- PMCID: PMC4904742
- DOI: 10.1038/srep27915
Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling
Abstract
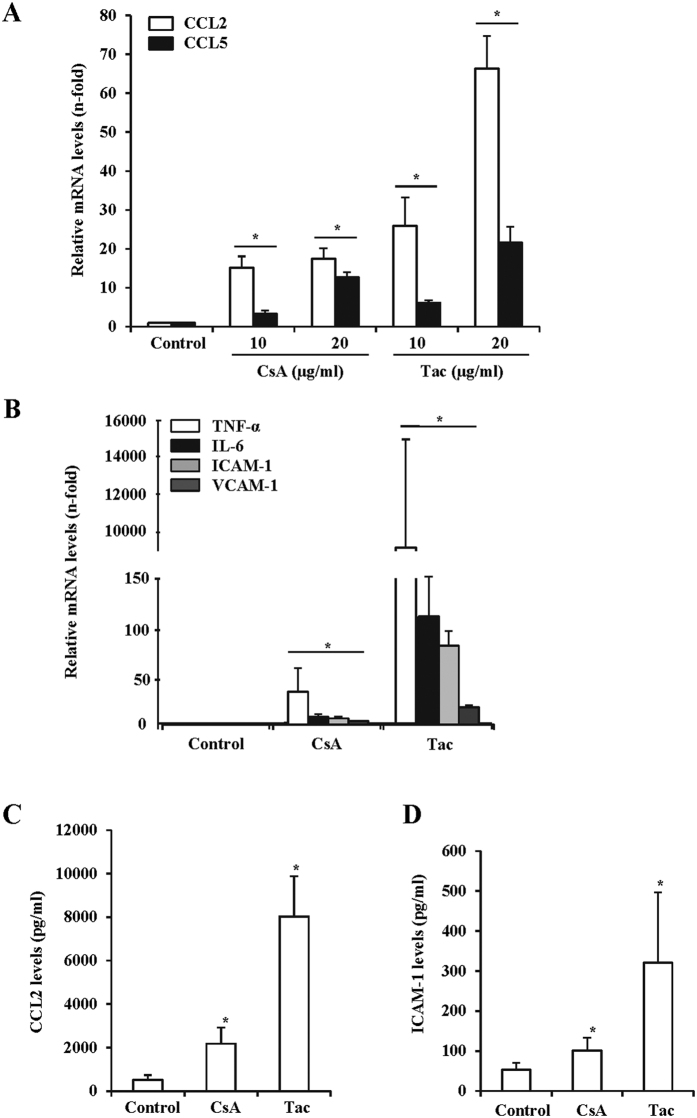
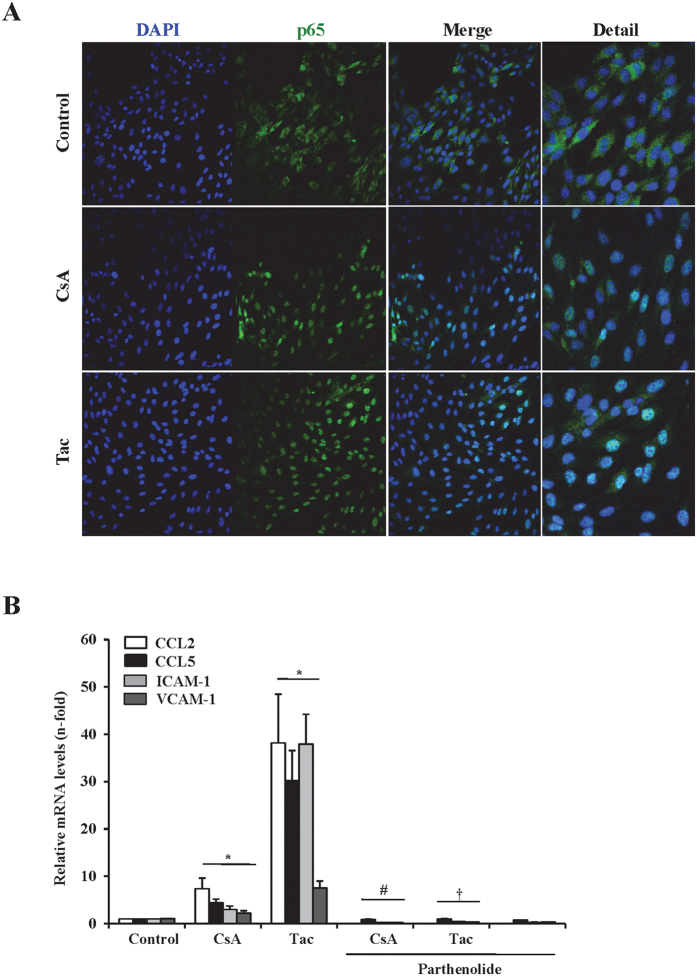
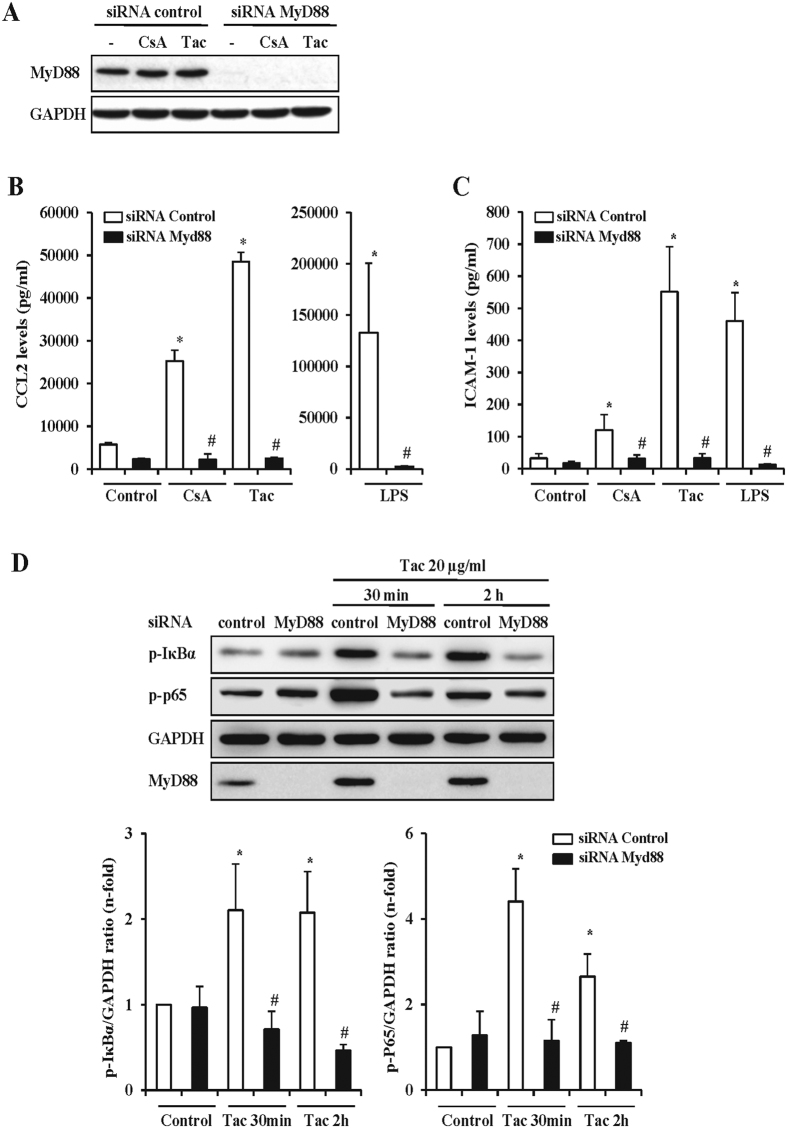
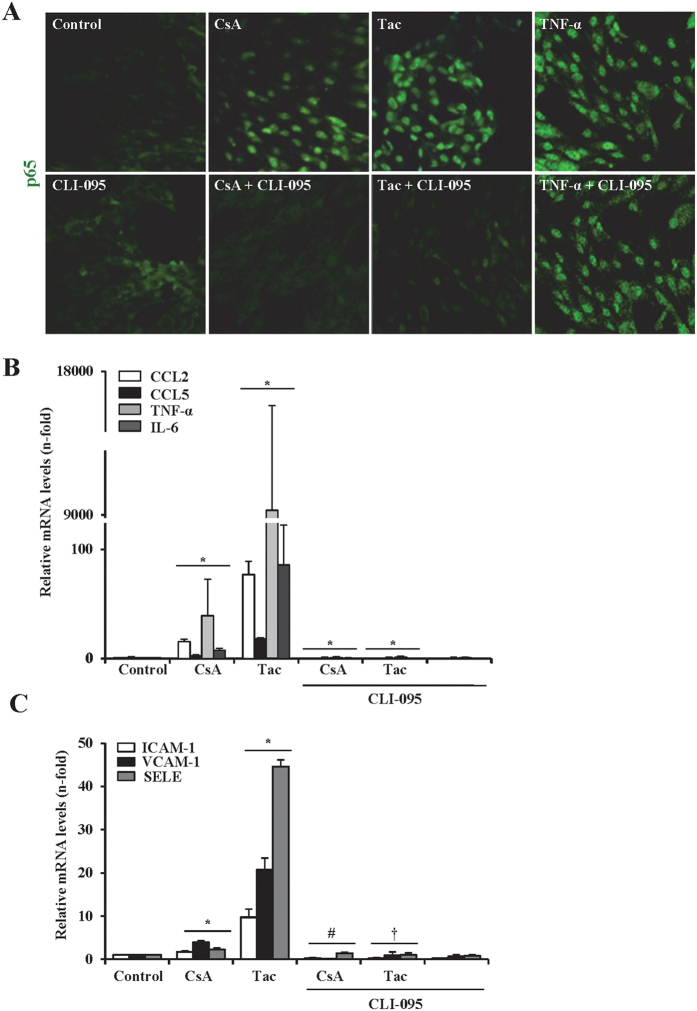
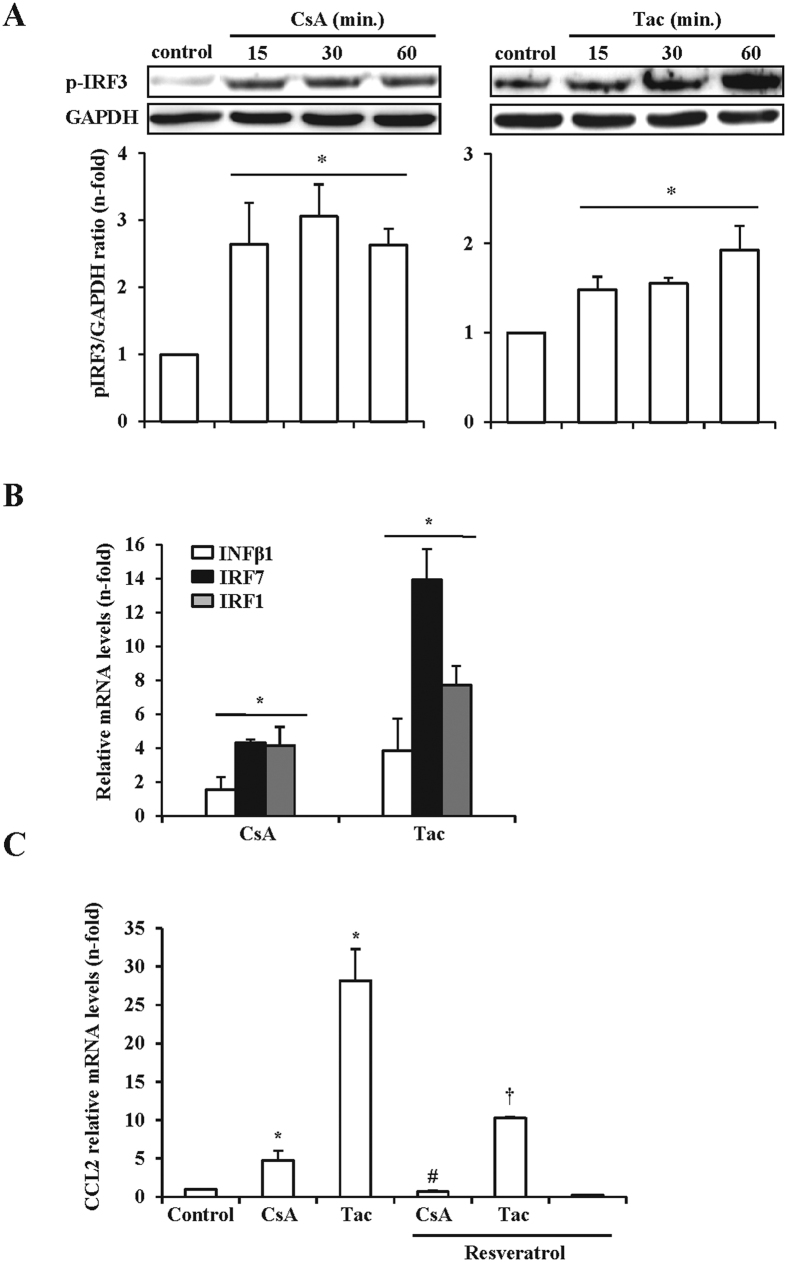
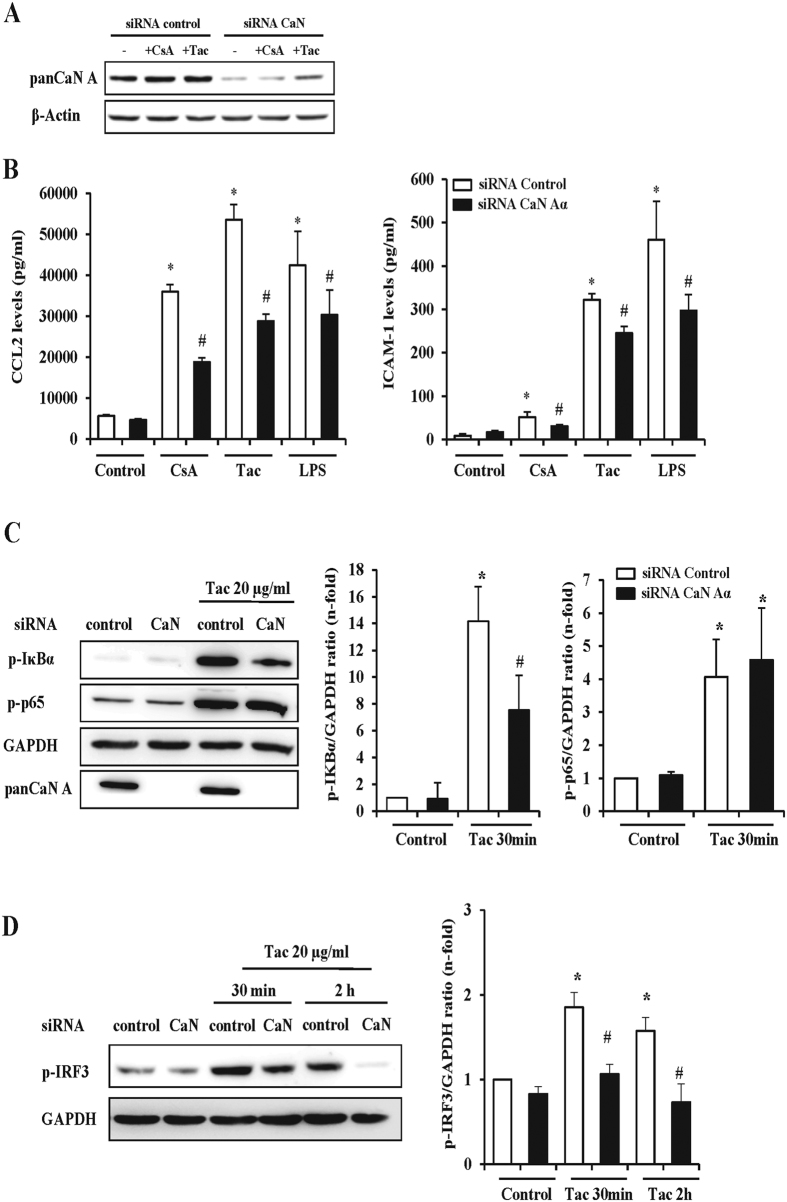
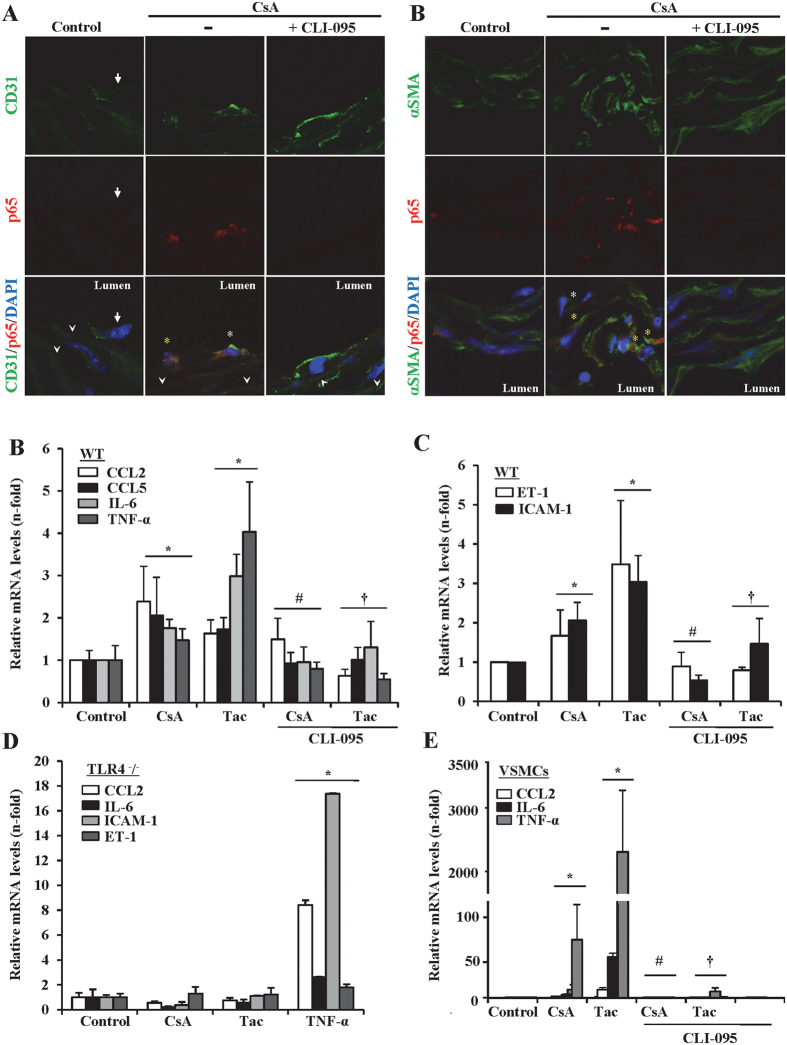
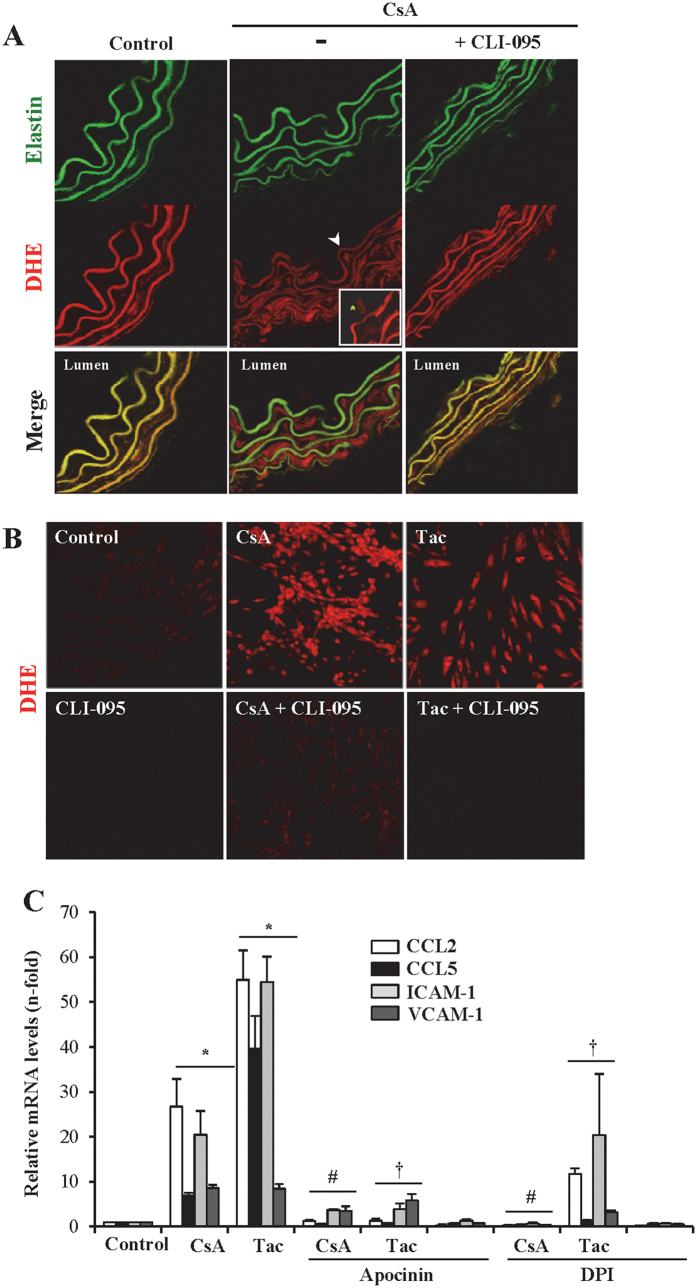
The introduction of the calcineurin inhibitors (CNIs) cyclosporine and tacrolimus greatly reduced the rate of allograft rejection, although their chronic use is marred by a range of side effects, among them vascular toxicity. In transplant patients, it is proved that innate immunity promotes vascular injury triggered by ischemia-reperfusion damage, atherosclerosis and hypertension. We hypothesized that activation of the innate immunity and inflammation may contribute to CNI toxicity, therefore we investigated whether TLR4 mediates toxic responses of CNIs in the vasculature. Cyclosporine and tacrolimus increased the production of proinflammatory cytokines and endothelial activation markers in cultured murine endothelial and vascular smooth muscle cells as well as in ex vivo cultures of murine aortas. CNI-induced proinflammatory events were prevented by pharmacological inhibition of TLR4. Moreover, CNIs were unable to induce inflammation and endothelial activation in aortas from TLR4(-/-) mice. CNI-induced cytokine and adhesion molecules synthesis in endothelial cells occurred even in the absence of calcineurin, although its expression was required for maximal effect through upregulation of TLR4 signaling. CNI-induced TLR4 activity increased O2(-)/ROS production and NF-κB-regulated synthesis of proinflammatory factors in cultured as well as aortic endothelial and VSMCs. These data provide new insight into the mechanisms associated with CNI vascular inflammation.
Figures
References
-
- Valantine H. Cardiac allograft vasculopathy after heart transplantation: risk factors and management. J Heart Lung Transplant. 23, S187–93 (2004). - PubMed
-
- Al-Massarani G. et al. Impact of immunosuppressive treatment on endothelial biomarkers after kidney transplantation. Am J Transplant. 8, 2360–7 (2008). - PubMed
-
- Mitchell R. N. Graft vascular disease: immune response meets the vessel wall. Annu Rev Pathol. 4, 19–47 (2009). - PubMed
-
- Navarro-Antolín J. et al. Role of peroxynitrite in endothelial damage mediated by Cyclosporine A. Free Radic Biol Med. 42, 394–403 (2007). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
