

FALDO: a semantic standard for describing the location of nucleotide and protein feature annotation
- PMID: 27296299
- PMCID: PMC4907002
- DOI: 10.1186/s13326-016-0067-z
FALDO: a semantic standard for describing the location of nucleotide and protein feature annotation
Abstract
Background: Nucleotide and protein sequence feature annotations are essential to understand biology on the genomic, transcriptomic, and proteomic level. Using Semantic Web technologies to query biological annotations, there was no standard that described this potentially complex location information as subject-predicate-object triples.
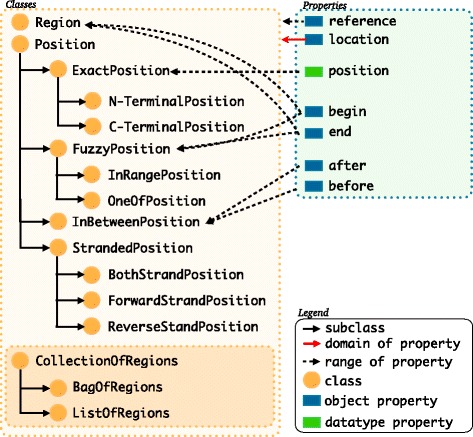
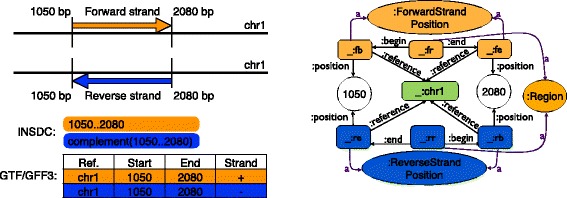
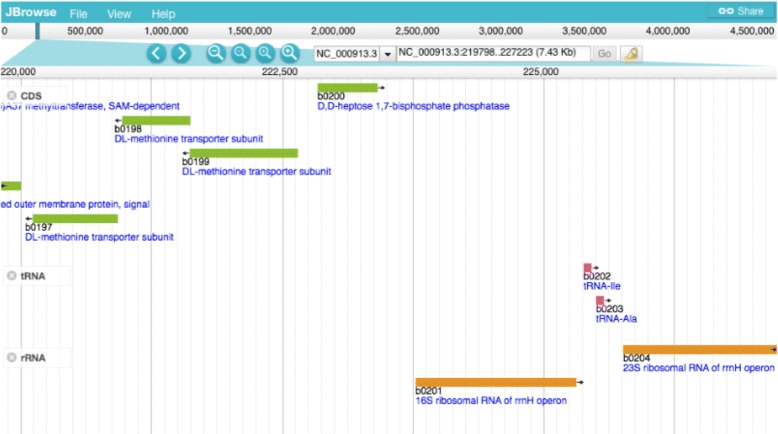
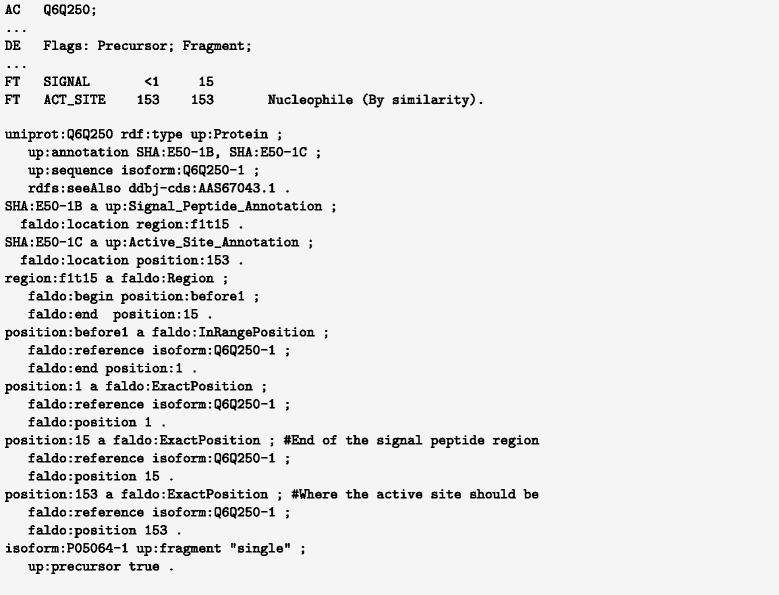
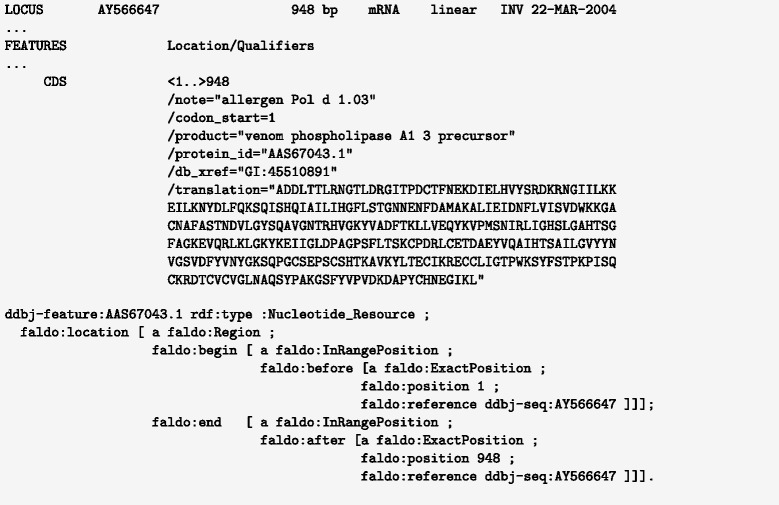
Description: We have developed an ontology, the Feature Annotation Location Description Ontology (FALDO), to describe the positions of annotated features on linear and circular sequences. FALDO can be used to describe nucleotide features in sequence records, protein annotations, and glycan binding sites, among other features in coordinate systems of the aforementioned "omics" areas. Using the same data format to represent sequence positions that are independent of file formats allows us to integrate sequence data from multiple sources and data types. The genome browser JBrowse is used to demonstrate accessing multiple SPARQL endpoints to display genomic feature annotations, as well as protein annotations from UniProt mapped to genomic locations.
Conclusions: Our ontology allows users to uniformly describe - and potentially merge - sequence annotations from multiple sources. Data sources using FALDO can prospectively be retrieved using federalised SPARQL queries against public SPARQL endpoints and/or local private triple stores.
Keywords: Annotation; Data integration; RDF; SPARQL; Semantic Web; Sequence feature; Sequence ontology; Standardisation.
Figures





References
-
- Dayhoff MO, Eck RV, Foundation NBR. Atlas of Protein Sequence and Structure. Silver Spring (Maryland): National Biomedical Research Foundation; 1965.
-
- Reese MG, Moore B, Batchelor C, Salas F, Cunningham F, Marth GT, Stein L, Flicek P, Yandell M, Eilbeck K. A standard variation file format for human genome sequences. Genome Biol. 2010; 11(R88). doi:10.1186/gb-2010-11-8-r88. - DOI - PMC - PubMed
-
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JGR, Korf I, Lapp H, Lehväslaiho H, Matsalla C, Mungall CJ, Osborne BI, Pocock MR, Schattner P, Senger M, Stein LD, Stupka E, Wilkinson MD, Birney E. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12(10):1611–8. doi: 10.1101/gr.361602. - DOI - PMC - PubMed
-
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25(11):1422–3. doi: 10.1093/bioinformatics/btp163. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
