

Pathogenesis and treatment of spine disease in the mucopolysaccharidoses
- PMID: 27296532
- PMCID: PMC4970936
- DOI: 10.1016/j.ymgme.2016.06.002
Pathogenesis and treatment of spine disease in the mucopolysaccharidoses
Abstract
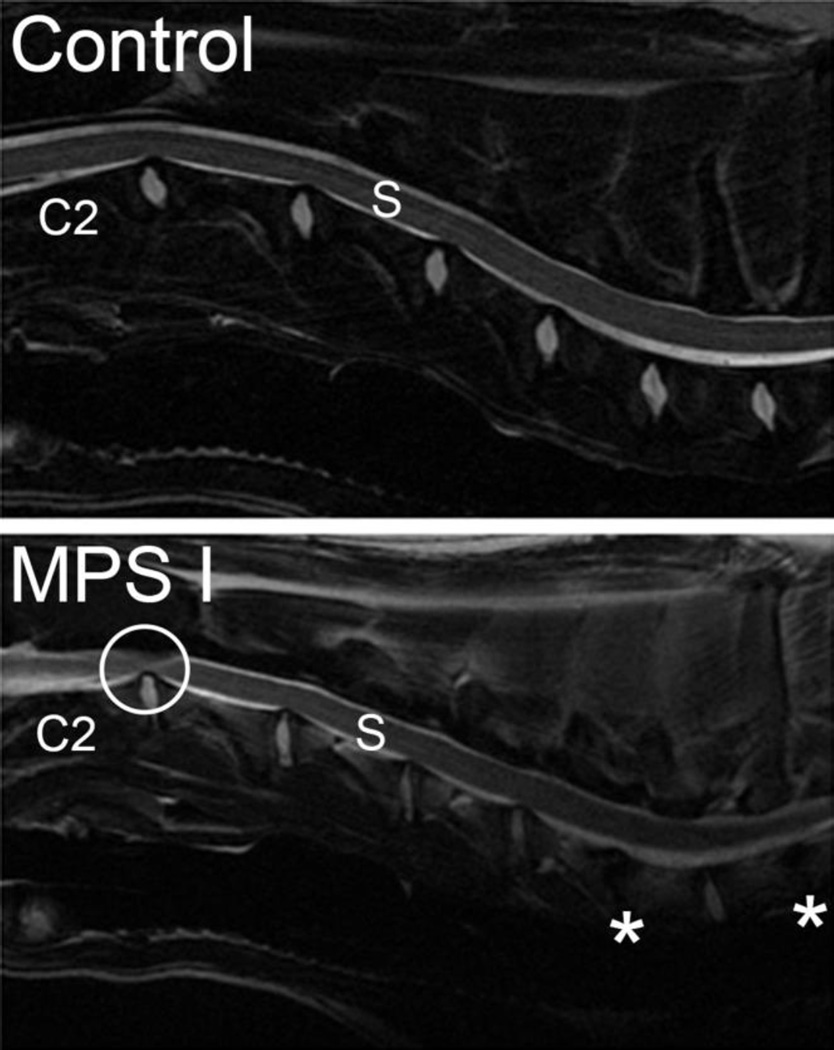
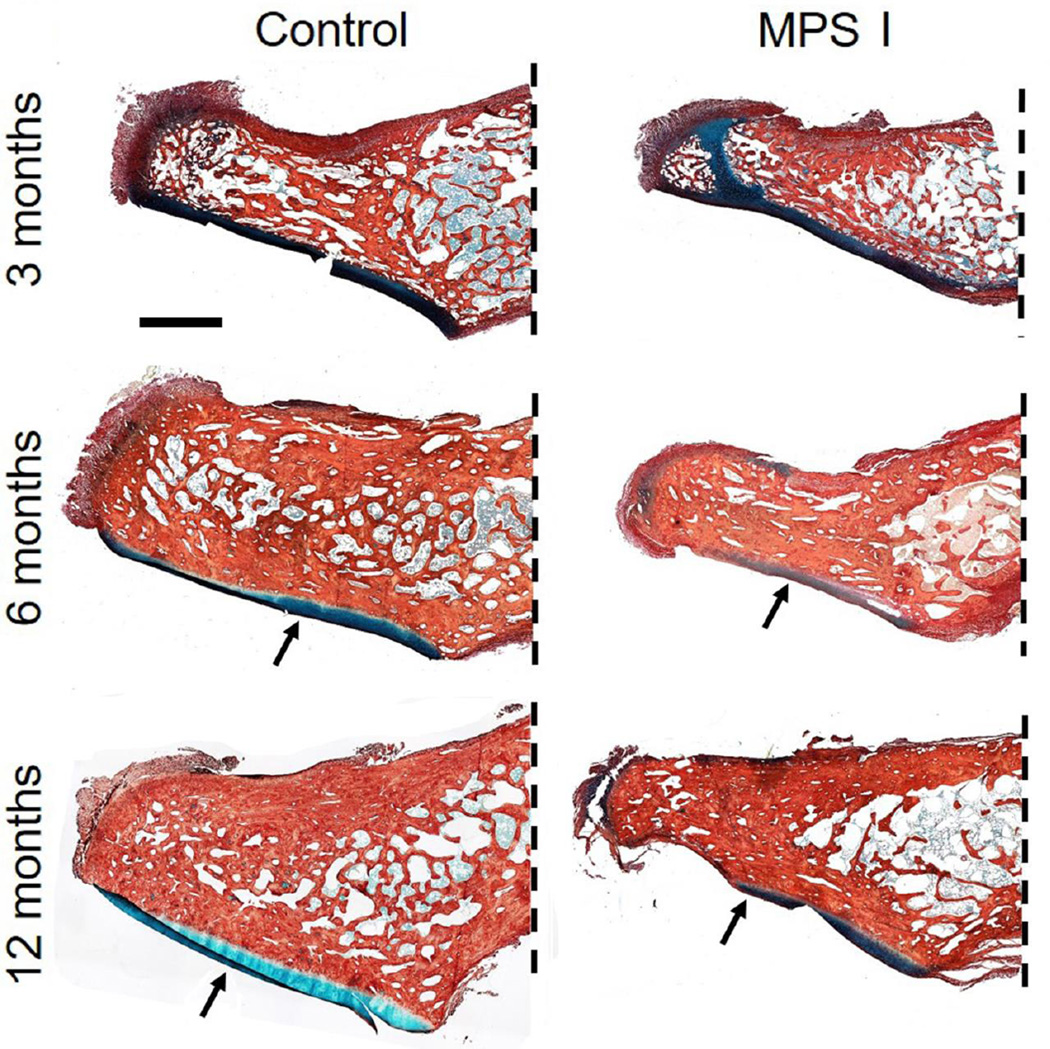
The mucopolysaccharidoses (MPS) are a family of lysosomal storage disorders characterized by deficient activity of enzymes that degrade glycosaminoglycans (GAGs). Skeletal disease is common in MPS patients, with the severity varying both within and between subtypes. Within the spectrum of skeletal disease, spinal manifestations are particularly prevalent. Developmental and degenerative abnormalities affecting the substructures of the spine can result in compression of the spinal cord and associated neural elements. Resulting neurological complications, including pain and paralysis, significantly reduce patient quality of life and life expectancy. Systemic therapies for MPS, such as hematopoietic stem cell transplantation and enzyme replacement therapy, have shown limited efficacy for improving spinal manifestations in patients and animal models. Therefore, there is a pressing need for new therapeutic approaches that specifically target this debilitating aspect of the disease. In this review, we examine how pathological abnormalities affecting the key substructures of the spine - the discs, vertebrae, odontoid process and dura - contribute to the progression of spinal deformity and symptomatic compression of neural elements. Specifically, we review current understanding of the underlying pathophysiology of spine disease in MPS, how the tissues of the spine respond to current clinical and experimental treatments, and discuss future strategies for improving the efficacy of these treatments.
Keywords: Animal models; Bone; Intervertebral disc; Lysosomal storage disorder; Mucopolysaccharidosis; Spine; Therapy; Vertebra.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures

References
-
- Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3421–3452.
-
- Wraith JE, Jones S. Mucopolysaccharidosis type I. Pediatr Endocrinol Rev. 2014;12(Suppl 1):102–106. - PubMed
-
- Esko JD, Kimata K, U L. Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2009. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
