

Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line
- PMID: 27303702
- PMCID: PMC4863610
- DOI: 10.1128/mSphere.00070-15
Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line
Abstract
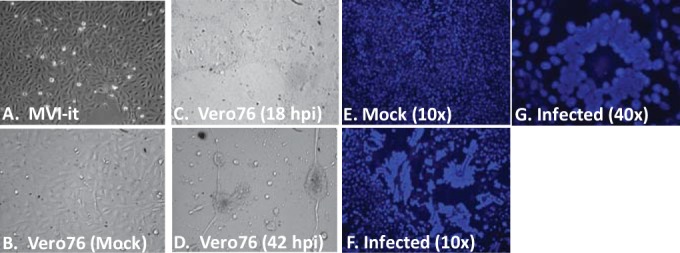
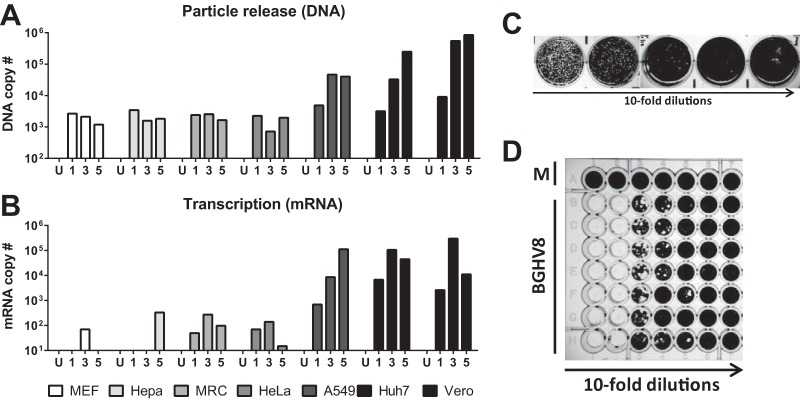
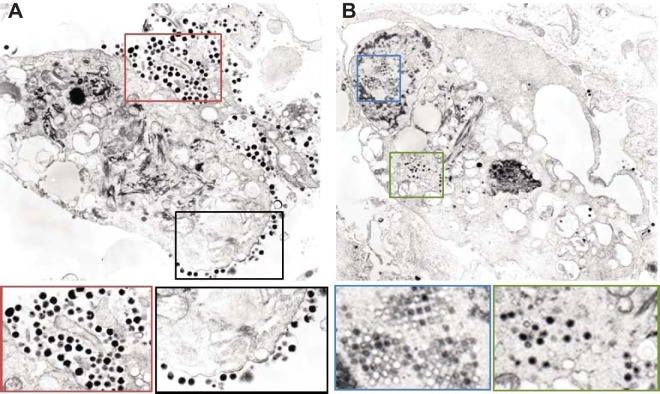
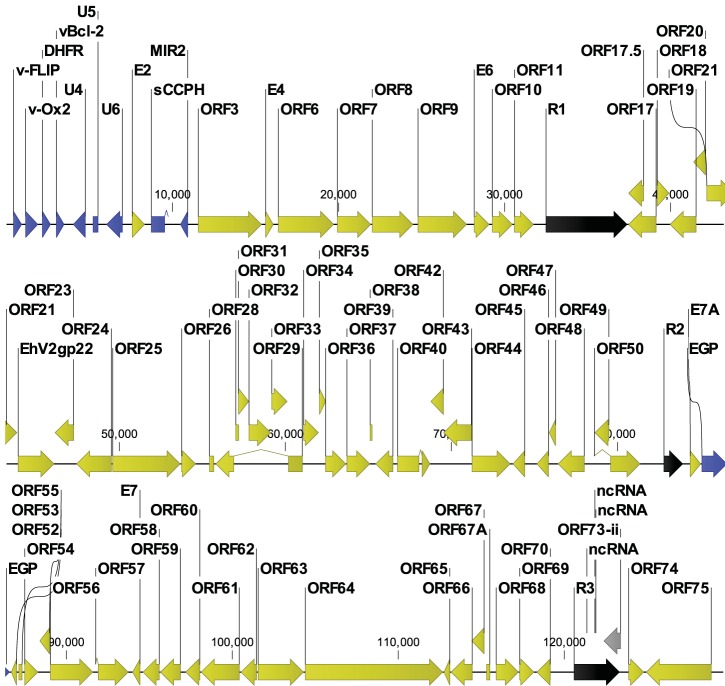
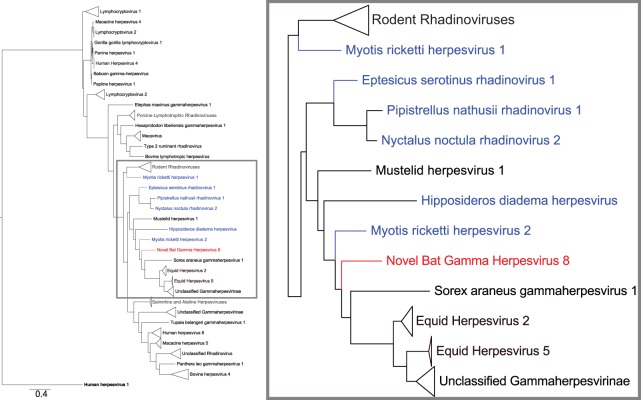
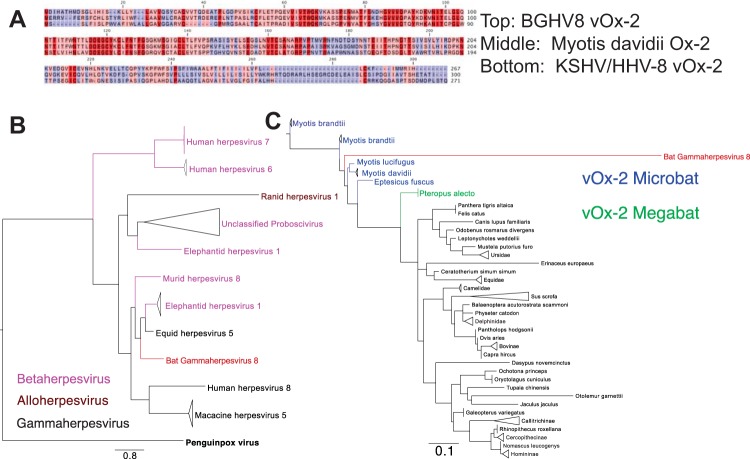
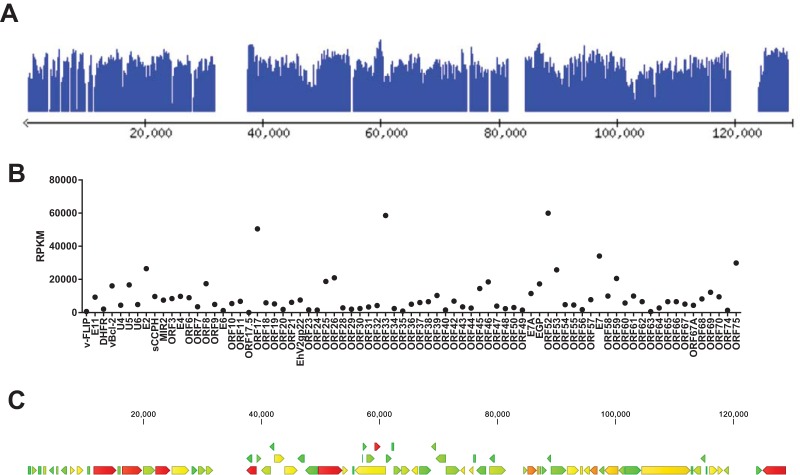
While employing deep sequencing and de novo assembly to characterize the mRNA transcript profile of a cell line derived from the microbat Myotis velifer incautus, we serendipitously identified mRNAs encoding proteins with a high level of identity to herpesviruses. A majority were closely related to proteins of equine herpesvirus 2 (EHV-2), a horse gammaherpesvirus. We demonstrated by electron microscopy the presence of herpesvirus-like particles in the microbat cells. Passage of supernatants from microbat cells to Vero cells resulted in syncytium formation, and expression of viral genes and amplification of viral DNA were demonstrated by quantitative PCR. Susceptibility of human cell lines to productive infection was also demonstrated. Next-generation sequencing and de novo assembly of the viral genome from supernatants from Vero cells yielded a single contig of approximately 130 kb with at least 77 open reading frames (ORFs), predicted microRNAs (miRNAs), and a gammaherpesvirus genomic organization. Phylogenic analysis of the envelope glycoprotein (gB) and DNA polymerase (POLD1) revealed similarity to multiple gammaherpesviruses, including those from as-yet-uncultured viruses of the Rhadinovirus genus that were obtained by deep sequencing of bat tissues. Moreover, the assembled genome revealed ORFs that share little or no homology to known ORFs in EHV-2 but are similar to accessory proteins of other gammaherpesviruses. Some also have striking homology to predicted Myotis bat proteins. Cumulatively, this study provides the first isolation and characterization of a replication-competent bat gammaherpesvirus. IMPORTANCE Bats are of significant interest as reservoirs for zoonotic viral pathogens; however, tools to dissect bat-virus interactions are limited in availability. This study serendipitously identified, in an established bat cell line, a fully replication-competent gammaherpesvirus; determined the complete genome sequence of the virus; and generated a viral transcript map. This virus can replicate in select human and nonhuman primate cell lines. However, analyses of viral sequences support a bat origin for this virus; we therefore refer to the virus as bat gammaherpesvirus 8 (BGHV8). The viral genome contains unique open reading frames that likely encode modulators of bat innate and adaptive immune signaling pathways and expresses viral miRNAs. The virus and its gene products should provide a unique tool to dissect both bat and gammaherpesvirus biology.
Keywords: bats; genomics; herpesviruses; next-generation sequencing; transcriptomics; virus discovery.
Figures
References
-
- Wang L-F, Cowled C. 2015. Bats and viruses: a new frontier of emerging infectious diseases. Wiley-Blackwell, Hoboken, NJ.
-
- Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, Djahanschiri B, Zeller G, Mende DR, Alberti A, Cornejo-Castillo FM, Costea PI, Cruaud C, d’Ovidio F, Engelen S, Ferrera I, Gasol JM, Guidi L, Hildebrand F, Kokoszka F, Lepoivre C, Lima-Mendez G, Poulain J, Poulos BT, Royo-Llonch M, Sarmento H, Vieira-Silva S, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Bowler C, de Vargas C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Jaillon O, Not F, Ogata H, Pesant S, Speich S, Stemmann L, Sullivan MB, Weissenbach J, Wincker P, Karsenti E, Raes J, Acinas SG, Bork P. 2015. Ocean plankton. Structure and function of the global ocean microbiome. Science 348:1261359. doi: 10.1126/science.1261359. - DOI - PubMed
-
- Bikel S, Valdez-Lara A, Cornejo-Granados F, Rico K, Canizales-Quinteros S, Soberon X, Del Pozo-Yauner L, Ochoa-Leyva A. 2015. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: towards a systems-level understanding of human microbiome. Comput Struct Biotechnol J 13:390–401. doi: 10.1016/j.csbj.2015.06.001. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
