

Active and Secretory IgA-Coated Bacterial Fractions Elucidate Dysbiosis in Clostridium difficile Infection
- PMID: 27303742
- PMCID: PMC4888886
- DOI: 10.1128/mSphere.00101-16
Active and Secretory IgA-Coated Bacterial Fractions Elucidate Dysbiosis in Clostridium difficile Infection
Abstract
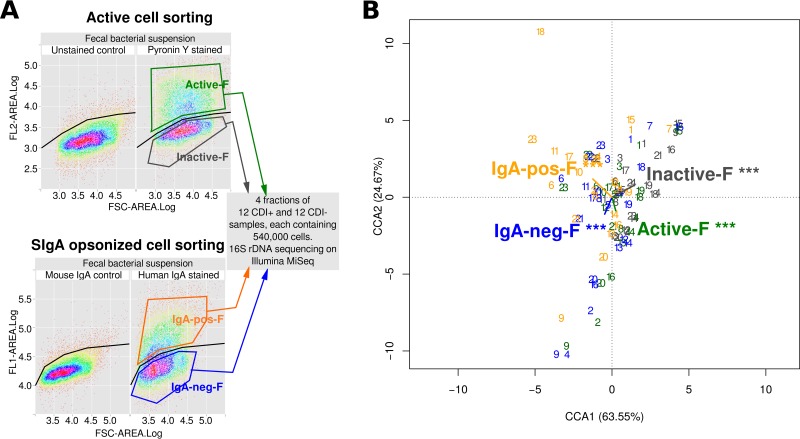
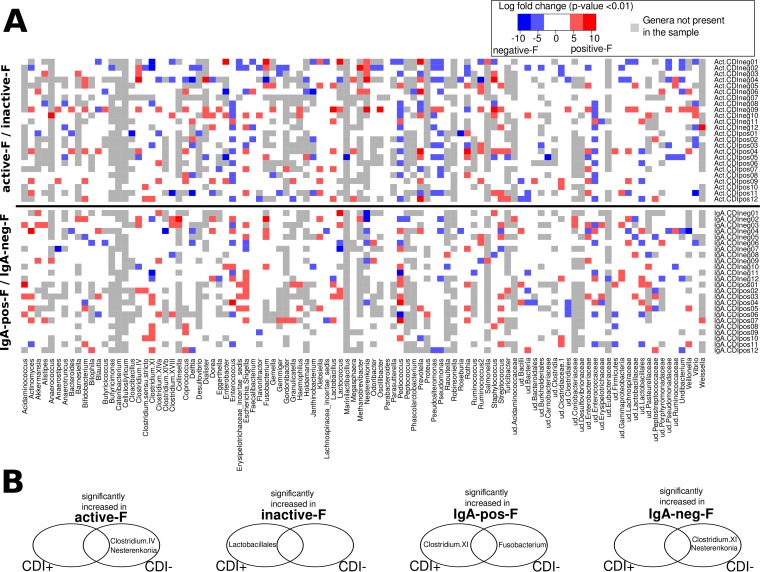
The onset of Clostridium difficile infection (CDI) has been associated with treatment with wide-spectrum antibiotics. Antibiotic treatment alters the activity of gut commensals and may result in modified patterns of immune responses to pathogens. To study these mechanisms during CDI, we separated bacteria with high cellular RNA content (the active bacteria) and their inactive counterparts by fluorescence-activated cell sorting (FACS) of the fecal bacterial suspension. The gut dysbiosis due to the antibiotic treatment may result in modification of immune recognition of intestinal bacteria. The immune recognition patterns were assessed by FACS of bacterial fractions either coated or not with intestinal secretory immunoglobulin A (SIgA). We described the taxonomic distributions of these four bacterial fractions (active versus inactive and SIgA coated versus non-SIgA coated) by massive 16S rRNA gene amplicon sequencing and quantified the proportion of C. difficile toxin genes in the samples. The overall gut microbiome composition was more robustly influenced by antibiotics than by the C. difficile toxins. Bayesian networks revealed that the C. difficile cluster was preferentially SIgA coated during CDI. In contrast, in the CDI-negative group Fusobacterium was the characteristic genus of the SIgA-opsonized fraction. Lactobacillales and Clostridium cluster IV were mostly inactive in CDI-positive patients. In conclusion, although the proportion of C. difficile in the gut is very low, it is able to initiate infection during the gut dysbiosis caused by environmental stress (antibiotic treatment) as a consequence of decreased activity of the protective bacteria. IMPORTANCE C. difficile is a major enteric pathogen with worldwide distribution. Its expansion is associated with broad-spectrum antibiotics which disturb the normal gut microbiome. In this study, the DNA sequencing of highly active bacteria and bacteria opsonized by intestinal secretory immunoglobulin A (SIgA) separated from the whole bacterial community by FACS elucidated how the gut dysbiosis promotes C. difficile infection (CDI). Bacterial groups with inhibitory effects on C. difficile growth, such as Lactobacillales, were mostly inactive in the CDI patients. C. difficile was typical for the bacterial fraction opsonized by SIgA in patients with CDI, while Fusobacterium was characteristic for the SIgA-opsonized fraction of the controls. The study demonstrates that sequencing of specific bacterial fractions provides additional information about dysbiotic processes in the gut. The detected patterns have been confirmed with the whole patient cohort independently of the taxonomic differences detected in the nonfractionated microbiomes.
Keywords: 16S rRNA gene sequencing; Bayesian networks; Clostridium difficile infection; antibiotics; dysbiosis; fluorescence-activated cell sorting; human gut microbiome; secretory immunoglobulin A.
Figures

Similar articles
-
Antibiotic Treatments for Clostridium difficile Infection Are Associated with Distinct Bacterial and Fungal Community Structures.mSphere. 2018 Jan 10;3(1):e00572-17. doi: 10.1128/mSphere.00572-17. eCollection 2018 Jan-Feb. mSphere. 2018. PMID: 29359185 Free PMC article.
-
Domestic canines do not display evidence of gut microbial dysbiosis in the presence of Clostridioides (Clostridium) difficile, despite cellular susceptibility to its toxins.Anaerobe. 2019 Aug;58:53-72. doi: 10.1016/j.anaerobe.2019.03.017. Epub 2019 Apr 1. Anaerobe. 2019. PMID: 30946985
-
Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization.Anaerobe. 2015 Aug;34:1-7. doi: 10.1016/j.anaerobe.2015.03.008. Epub 2015 Mar 26. Anaerobe. 2015. PMID: 25817005
-
Insights into the Interaction Between Clostridioides difficile and the Gut Microbiome.J Pers Med. 2025 Feb 28;15(3):94. doi: 10.3390/jpm15030094. J Pers Med. 2025. PMID: 40137411 Free PMC article. Review.
-
Breakthroughs in the treatment and prevention of Clostridium difficile infection.Nat Rev Gastroenterol Hepatol. 2016 Mar;13(3):150-60. doi: 10.1038/nrgastro.2015.220. Epub 2016 Feb 10. Nat Rev Gastroenterol Hepatol. 2016. PMID: 26860266 Review.
Cited by
-
Engineered Secretory Immunoglobulin A provides insights on antibody-based effector mechanisms targeting Clostridiodes difficile.bioRxiv [Preprint]. 2023 Nov 12:2023.11.08.566291. doi: 10.1101/2023.11.08.566291. bioRxiv. 2023. PMID: 37986930 Free PMC article. Preprint.
-
Oxidative stress in the oral cavity is driven by individual-specific bacterial communities.NPJ Biofilms Microbiomes. 2018 Nov 27;4:29. doi: 10.1038/s41522-018-0072-3. eCollection 2018. NPJ Biofilms Microbiomes. 2018. PMID: 30510769 Free PMC article.
-
Oral Administration of Compound Probiotics Improved Canine Feed Intake, Weight Gain, Immunity and Intestinal Microbiota.Front Immunol. 2019 Apr 2;10:666. doi: 10.3389/fimmu.2019.00666. eCollection 2019. Front Immunol. 2019. PMID: 31001271 Free PMC article.
-
Characterization of intestinal microbiota and fecal cortisol, T3, and IgA in forest musk deer (Moschus berezovskii) from birth to weaning.Integr Zool. 2021 May;16(3):300-312. doi: 10.1111/1749-4877.12522. Epub 2021 Jan 22. Integr Zool. 2021. PMID: 33452844 Free PMC article.
-
Detection of mixed-strain infections by FACS and ultra-low input genome sequencing.Gut Microbes. 2020 May 3;11(3):305-309. doi: 10.1080/19490976.2018.1526578. Epub 2018 Oct 5. Gut Microbes. 2020. PMID: 30289342 Free PMC article.
References
-
- Rojo D, Gosalbes MJ, Ferrari R, Pérez-Cobas AE, Hernández E, Oltra R, Buesa J, Latorre A, Barbas C, Ferrer M, Moya A. 2015. Clostridium difficile heterogeneously impacts intestinal community architecture but drives stable metabolome responses. ISME J 9:2206–2220. doi:10.1038/ismej.2015.32. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
