

Crucial factors of the inflammatory microenvironment (IL-1β/TNF-α/TIMP-1) promote the maintenance of the malignant hemopoietic clone of myelofibrosis: an in vitro study
- PMID: 27304059
- PMCID: PMC5190072
- DOI: 10.18632/oncotarget.9949
Crucial factors of the inflammatory microenvironment (IL-1β/TNF-α/TIMP-1) promote the maintenance of the malignant hemopoietic clone of myelofibrosis: an in vitro study
Abstract
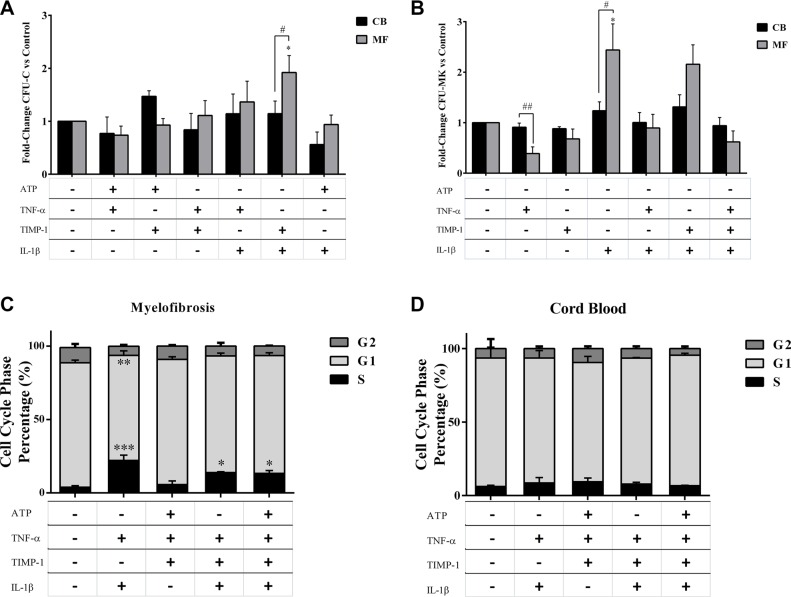
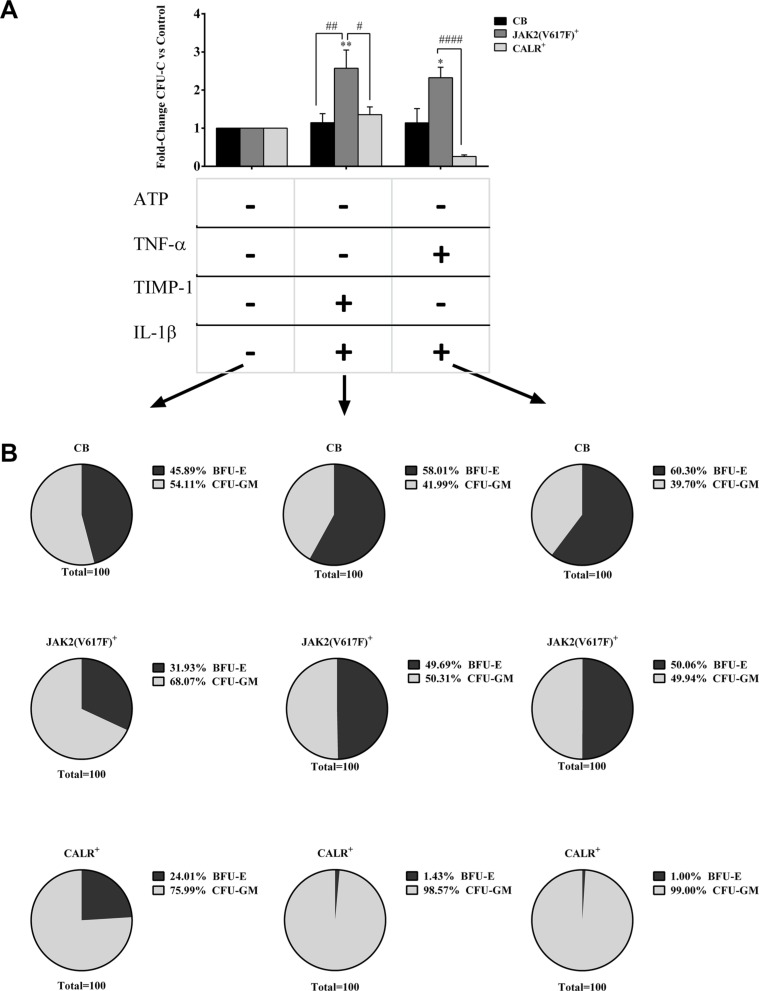
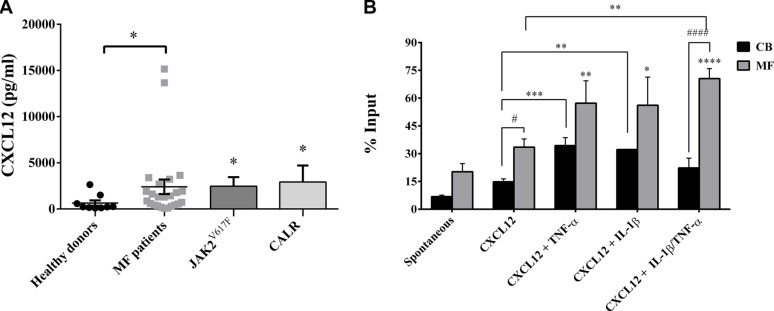
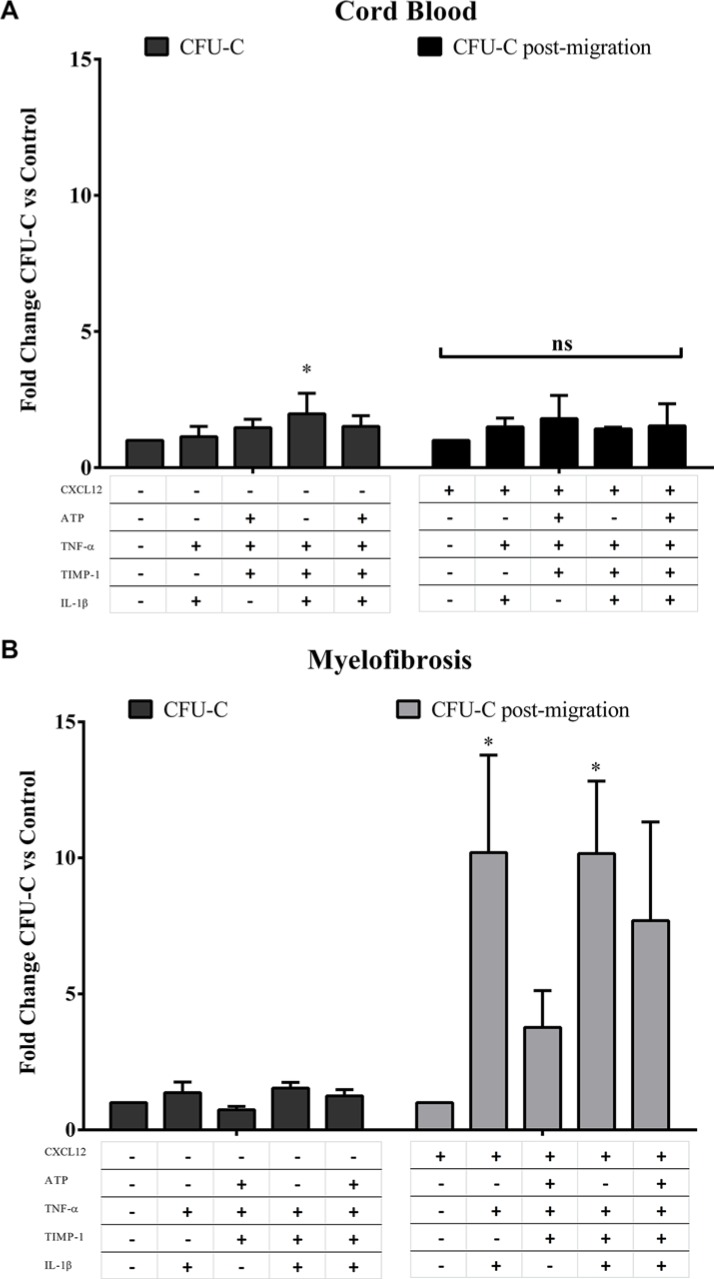
Along with molecular abnormalities (mutations in JAK2, Calreticulin (CALR) and MPL genes), chronic inflammation is the major hallmark of Myelofibrosis (MF). Here, we investigated the in vitro effects of crucial factors of the inflammatory microenvironment (Interleukin (IL)-1β, Tumor Necrosis Factor (TNF)-α, Tissue Inhibitor of Metalloproteinases (TIMP)-1 and ATP) on the functional behaviour of MF-derived circulating CD34+ cells.We found that, regardless mutation status, IL-1β or TNF-α increases the survival of MF-derived CD34+ cells. In addition, along with stimulation of cell cycle progression to the S-phase, IL-1β or TNF-α ± TIMP-1 significantly stimulate(s) the in vitro clonogenic ability of CD34+ cells from JAK2V617 mutated patients. Whereas in the JAK2V617F mutated group, the addition of IL-1β or TNF-α + TIMP-1 decreased the erythroid compartment of the CALR mutated patients. Megakaryocyte progenitors were stimulated by IL-1β (JAK2V617F mutated patients only) and inhibited by TNF-α. IL-1β + TNF-α + C-X-C motif chemokine 12 (CXCL12) ± TIMP-1 highly stimulates the in vitro migration of MF-derived CD34+ cells. Interestingly, after migration toward IL-1β + TNF-α + CXCL12 ± TIMP-1, CD34+ cells from JAK2V617F mutated patients show increased clonogenic ability.Here we demonstrate that the interplay of these inflammatory factors promotes and selects the circulating MF-derived CD34+ cells with higher proliferative activity, clonogenic potential and migration ability. Targeting these micro-environmental interactions may be a clinically relevant approach.
Keywords: circulating CD34+ cells; inflammatory microenvironment; migration; myelofibrosis; survival.
Conflict of interest statement
The Authors declare no conflicts of interest.
Figures



Similar articles
-
Distinct profile of CD34+ cells and plasma-derived extracellular vesicles from triple-negative patients with Myelofibrosis reveals potential markers of aggressive disease.J Exp Clin Cancer Res. 2021 Feb 1;40(1):49. doi: 10.1186/s13046-020-01776-8. J Exp Clin Cancer Res. 2021. PMID: 33522952 Free PMC article.
-
Mobilized Peripheral Blood versus Cord Blood: Insight into the Distinct Role of Proinflammatory Cytokines on Survival, Clonogenic Ability, and Migration of CD34+ Cells.Mediators Inflamm. 2018 Jul 4;2018:5974613. doi: 10.1155/2018/5974613. eCollection 2018. Mediators Inflamm. 2018. PMID: 30116149 Free PMC article.
-
Pro-inflammatory IL-1beta and/or TNF-alpha up-regulate matrix metalloproteases-1 and -3 mRNA in chondrocyte subpopulations potentially pathogenic in osteoarthritis: in situ hybridization studies on a single cell level.Int J Rheum Dis. 2016 Jun;19(6):557-66. doi: 10.1111/1756-185X.12431. Epub 2014 Oct 8. Int J Rheum Dis. 2016. PMID: 25291965
-
A thrombopoietin receptor antagonist is capable of depleting myelofibrosis hematopoietic stem and progenitor cells.Blood. 2016 Jun 30;127(26):3398-409. doi: 10.1182/blood-2015-10-674465. Epub 2016 Apr 25. Blood. 2016. PMID: 27114459 Free PMC article.
-
Folate metabolism in myelofibrosis: a missing key?Ann Hematol. 2025 Jan;104(1):35-46. doi: 10.1007/s00277-024-06176-y. Epub 2025 Jan 23. Ann Hematol. 2025. PMID: 39847116 Free PMC article. Review.
Cited by
-
Putative associations between inflammatory biomarkers, obesity, and obstructive sleep apnea.Ann Thorac Med. 2021 Oct-Dec;16(4):329-336. doi: 10.4103/atm.atm_644_20. Epub 2021 Sep 15. Ann Thorac Med. 2021. PMID: 34820020 Free PMC article.
-
Spatial and biochemical interactions between bone marrow adipose tissue and hematopoietic stem and progenitor cells in rhesus macaques.Bone. 2020 Apr;133:115248. doi: 10.1016/j.bone.2020.115248. Epub 2020 Jan 20. Bone. 2020. PMID: 31972314 Free PMC article.
-
Early Plasma Matrix Metalloproteinase Profiles. A Novel Pathway in Pediatric Acute Respiratory Distress Syndrome.Am J Respir Crit Care Med. 2019 Jan 15;199(2):181-189. doi: 10.1164/rccm.201804-0678OC. Am J Respir Crit Care Med. 2019. PMID: 30114376 Free PMC article.
-
The role of the extracellular matrix in primary myelofibrosis.Blood Cancer J. 2017 Feb 3;7(2):e525. doi: 10.1038/bcj.2017.6. Blood Cancer J. 2017. PMID: 28157219 Free PMC article. Review.
-
Anti-inflammatory treatment in MPN: targeting TNFR1 and TNFR2 in JAK2-V617F-induced disease.Blood Adv. 2021 Dec 14;5(23):5349-5359. doi: 10.1182/bloodadvances.2021004438. Blood Adv. 2021. PMID: 34592754 Free PMC article.
References
-
- Kleppe M, Levine RL. New pieces of a puzzle: the current biological picture of MPN. Biochim Biophys Acta. 2012;1826:415–22. - PubMed
-
- Tefferi A, Pardanani A. Myeloproliferative Neoplasms: a contemporary review. JAMA Oncol. 2015;1:97–105. - PubMed
-
- James C, Ugo V, Casadevall N, Constantinescu SN, Vainchenker W. A JAK2 mutation in myeloproliferative disorders : pathogenesis and therapeutic and scientific prospects. Trend Mol Med. 2005;11:546–54. - PubMed
-
- Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding and treatment strategies. J Clin Oncol. 2011;29:573–82. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
