

From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence
- PMID: 27304503
- PMCID: PMC4911819
- DOI: 10.1016/j.cmet.2016.05.010
From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence
Abstract
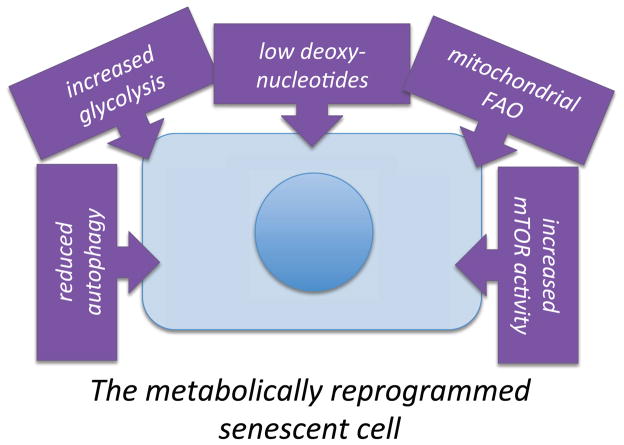
Cellular senescence is a complex stress response that permanently arrests the proliferation of cells at risk for oncogenic transformation. However, senescent cells can also drive phenotypes associated with aging. Although the senescence-associated growth arrest prevents the development of cancer, and the metabolism of cancer cells has been studied in depth, the metabolic causes and consequences of cellular senescence were largely unexplored until recently. New findings reveal key roles for several aspects of cellular metabolism in the establishment and control of senescent phenotypes. These discoveries have important implications for both cancer and aging. In this review, we highlight some of the recent links between metabolism and phenotypes that are commonly associated with senescent cells.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures


References
-
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. - PubMed
-
- Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. - PubMed
-
- Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, Kumar R, Jenkins RB, de Groen PC, Roche P, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet. 2004;36:744–749. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
