

Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction
- PMID: 27307396
- PMCID: PMC4988286
- DOI: 10.1111/jcmm.12893
Methylglyoxal induces cell death through endoplasmic reticulum stress-associated ROS production and mitochondrial dysfunction
Abstract
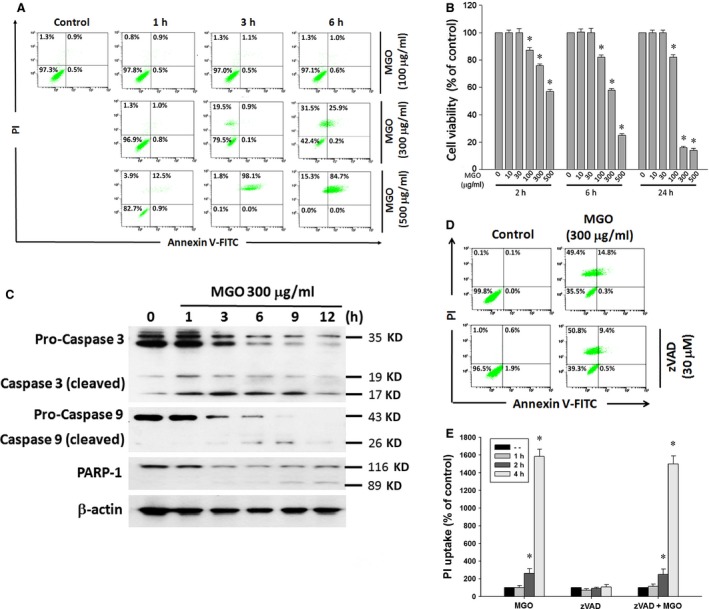
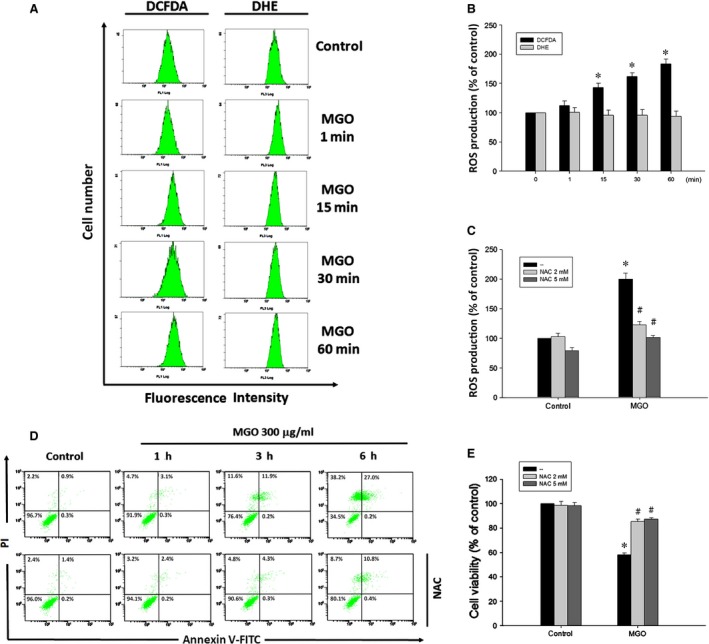
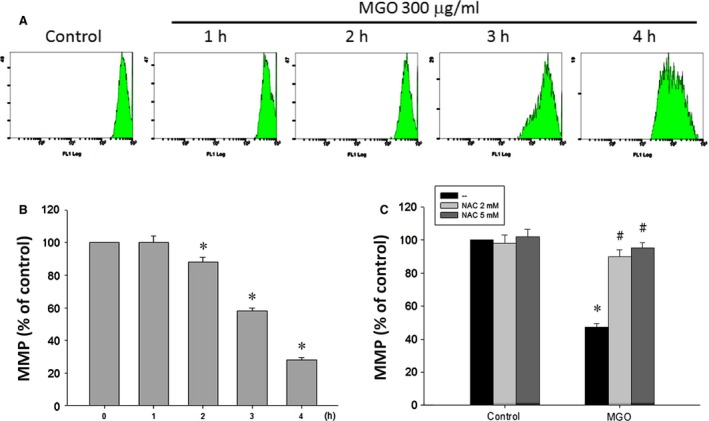
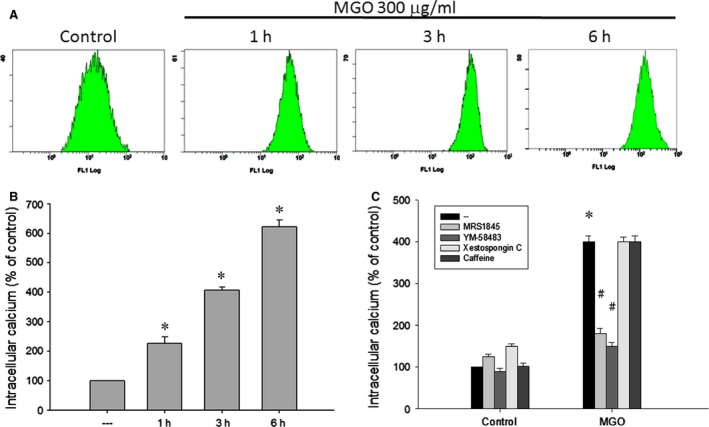
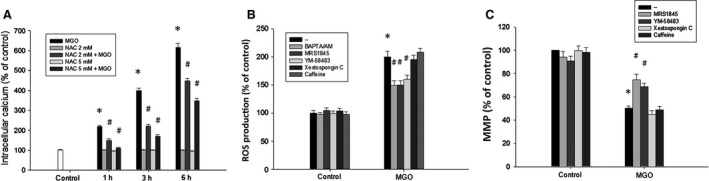
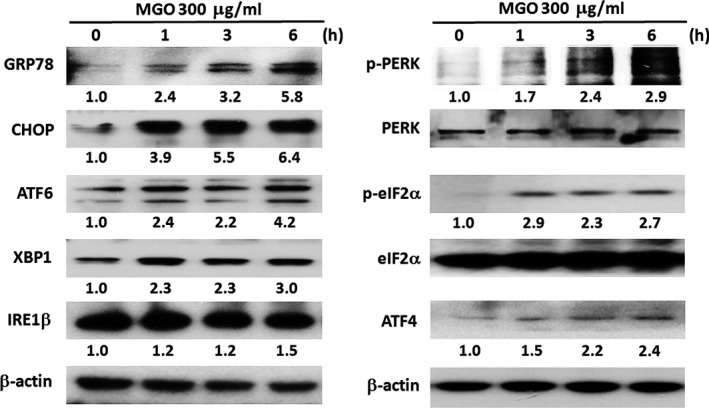
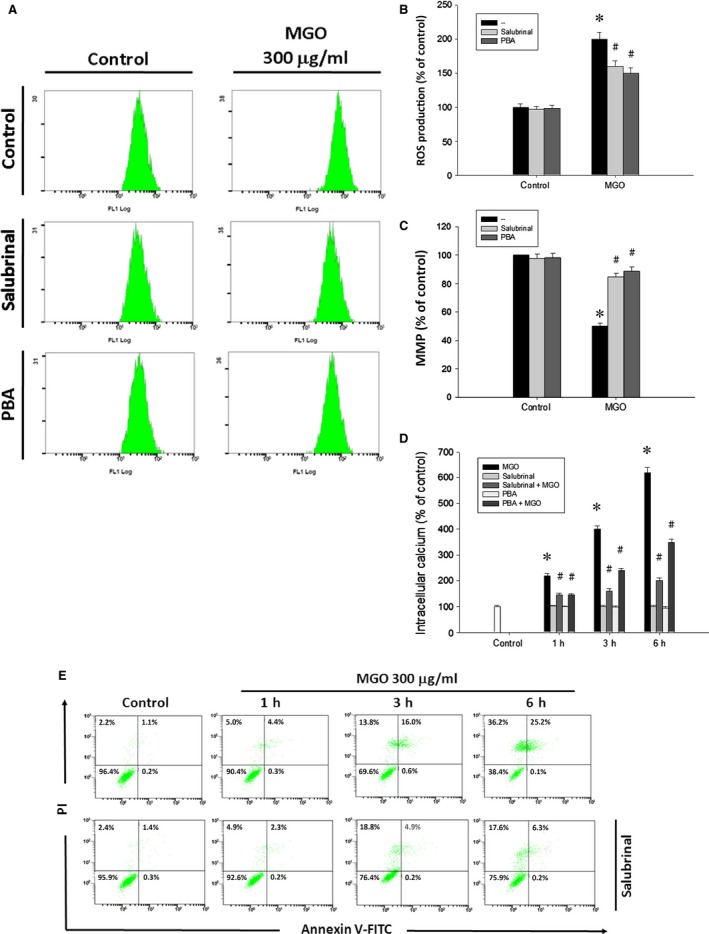
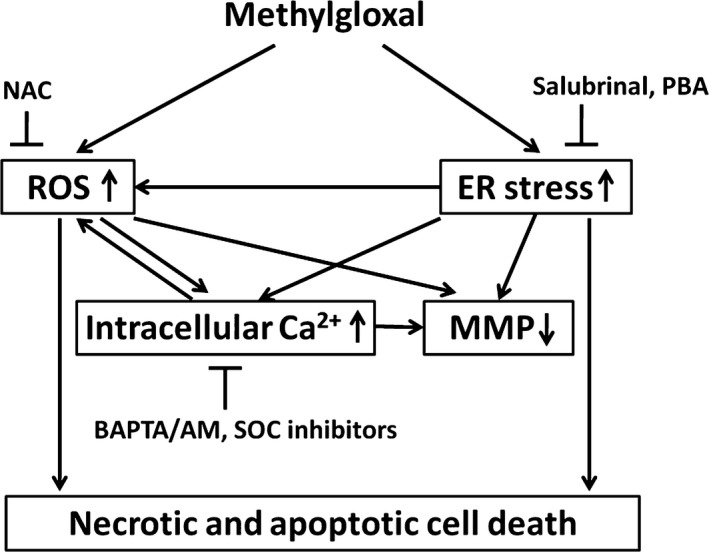
Diabetic retinopathy (DR) and age-related macular degeneration (AMD) are two important leading causes of acquired blindness in developed countries. As accumulation of advanced glycation end products (AGEs) in retinal pigment epithelial (RPE) cells plays an important role in both DR and AMD, and the methylglyoxal (MGO) within the AGEs exerts irreversible effects on protein structure and function, it is crucial to understand the underlying mechanism of MGO-induced RPE cell death. Using ARPE-19 as the cell model, this study revealed that MGO induces RPE cell death through a caspase-independent manner, which relying on reactive oxygen species (ROS) formation, mitochondrial membrane potential (MMP) loss, intracellular calcium elevation and endoplasmic reticulum (ER) stress response. Suppression of ROS generation can reverse the MGO-induced ROS production, MMP loss, intracellular calcium increase and cell death. Moreover, store-operated calcium channel inhibitors MRS1845 and YM-58483, but not the inositol 1,4,5-trisphosphate (IP3) receptor inhibitor xestospongin C, can block MGO-induced ROS production, MMP loss and sustained intracellular calcium increase in ARPE-19 cells. Lastly, inhibition of ER stress by salubrinal and 4-PBA can reduce the MGO-induced intracellular events and cell death. Therefore, our data indicate that MGO can decrease RPE cell viability, resulting from the ER stress-dependent intracellular ROS production, MMP loss and increased intracellular calcium increase. As MGO is one of the components of drusen in AMD and is the AGEs adduct in DR, this study could provide a valuable insight into the molecular pathogenesis and therapeutic intervention of AMD and DR.
Keywords: ER stress; intracellular calcium; methylglyoxal; mitochondria; reactive oxygen species; retinal pigment epithelium.
© 2016 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
Figures
References
-
- Saaddine JB, Honeycutt AA, Narayan KM, et al Projection of diabetic retinopathy and other major eye diseases among people with diabetes mellitus: United States, 2005‐2050. Arch Ophthalmol. 2008; 126: 1740–7. - PubMed
-
- Congdon N, O'Colmain B, Klaver CC, et al Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004; 122: 477–85. - PubMed
-
- Mettu PS, Wielgus AR, Ong SS, et al Retinal pigment epithelium response to oxidant injury in the pathogenesis of early age‐related macular degeneration. Mol Aspects Med. 2012; 33: 376–98. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
