

Proteome-wide covalent ligand discovery in native biological systems
- PMID: 27309814
- PMCID: PMC4919207
- DOI: 10.1038/nature18002
Proteome-wide covalent ligand discovery in native biological systems
Abstract
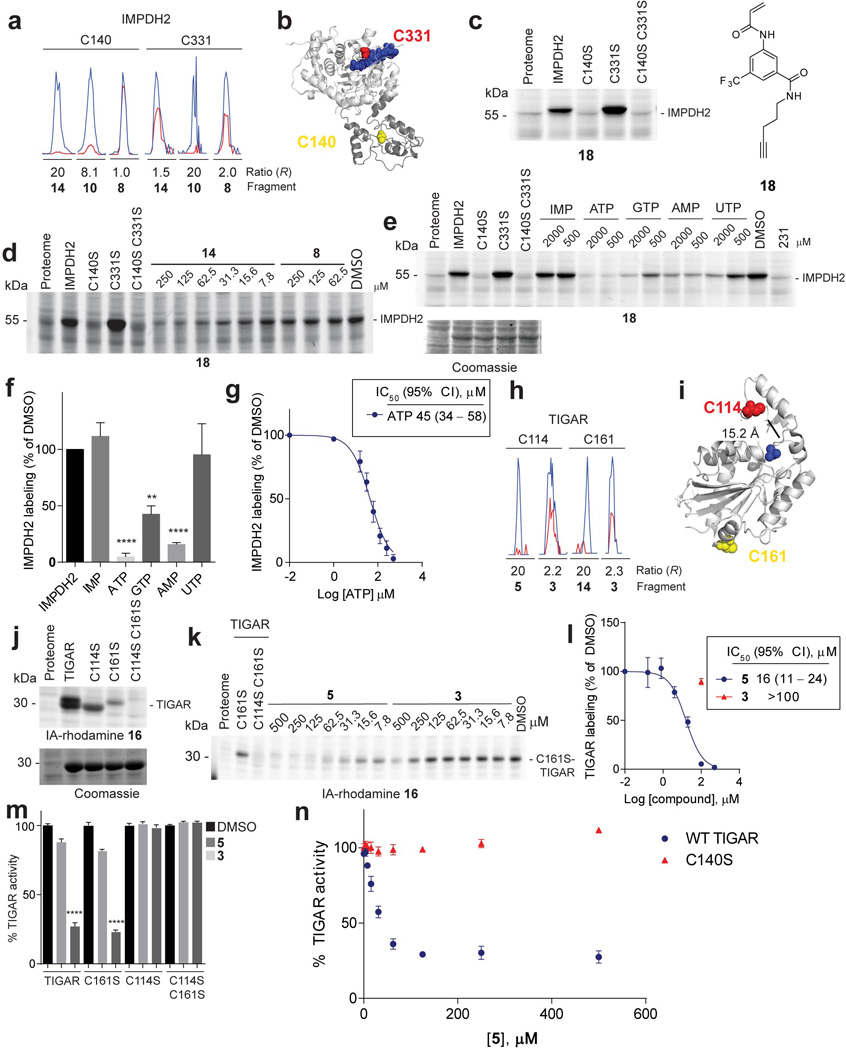
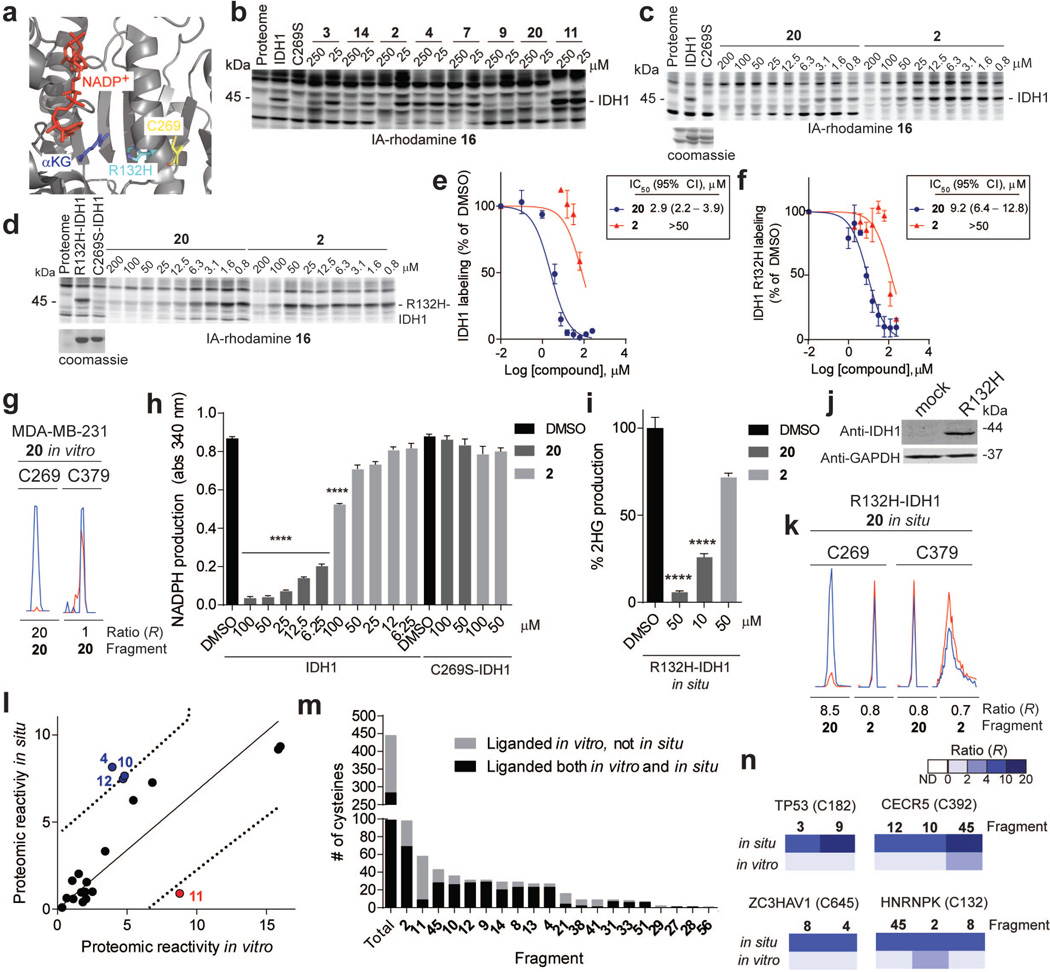
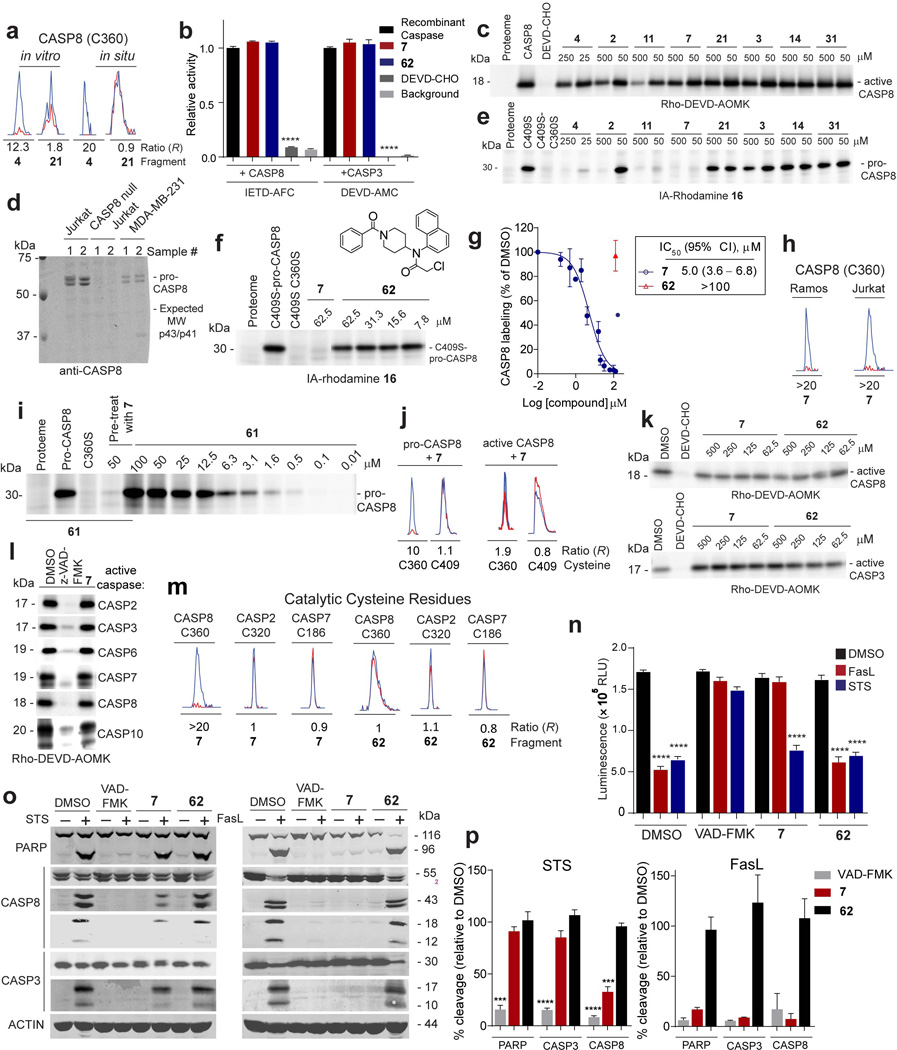
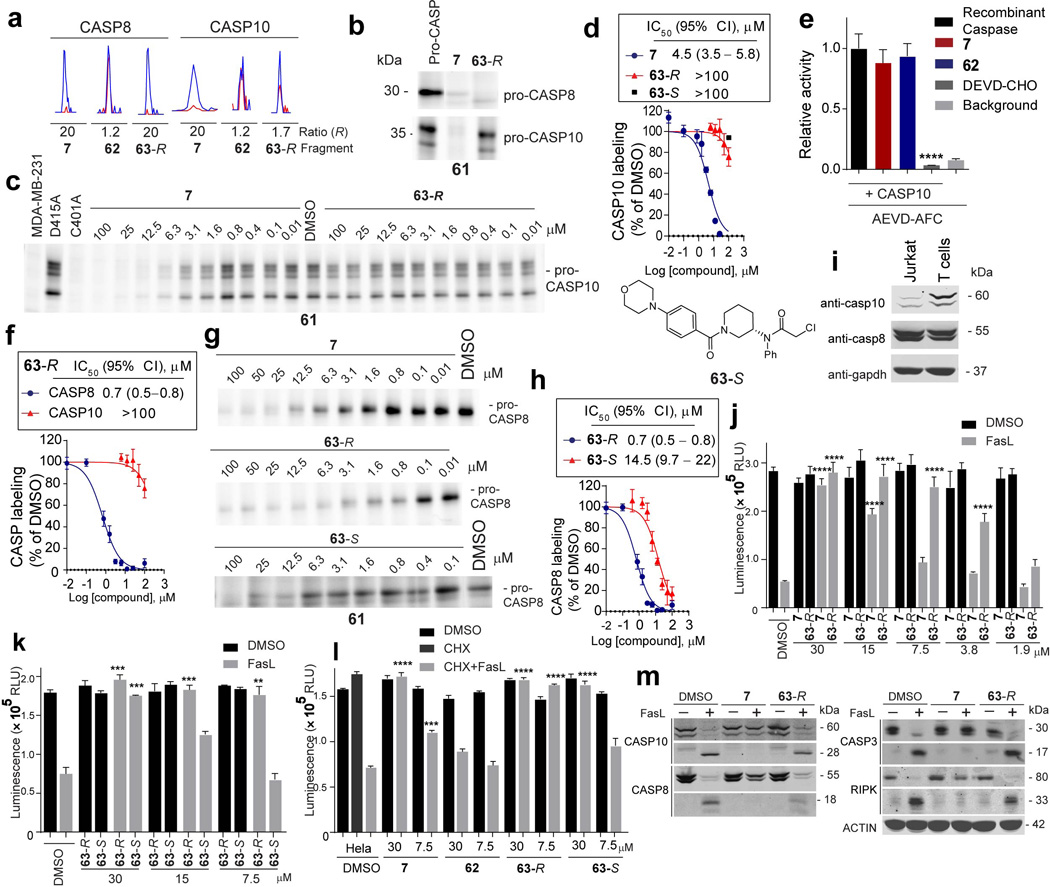
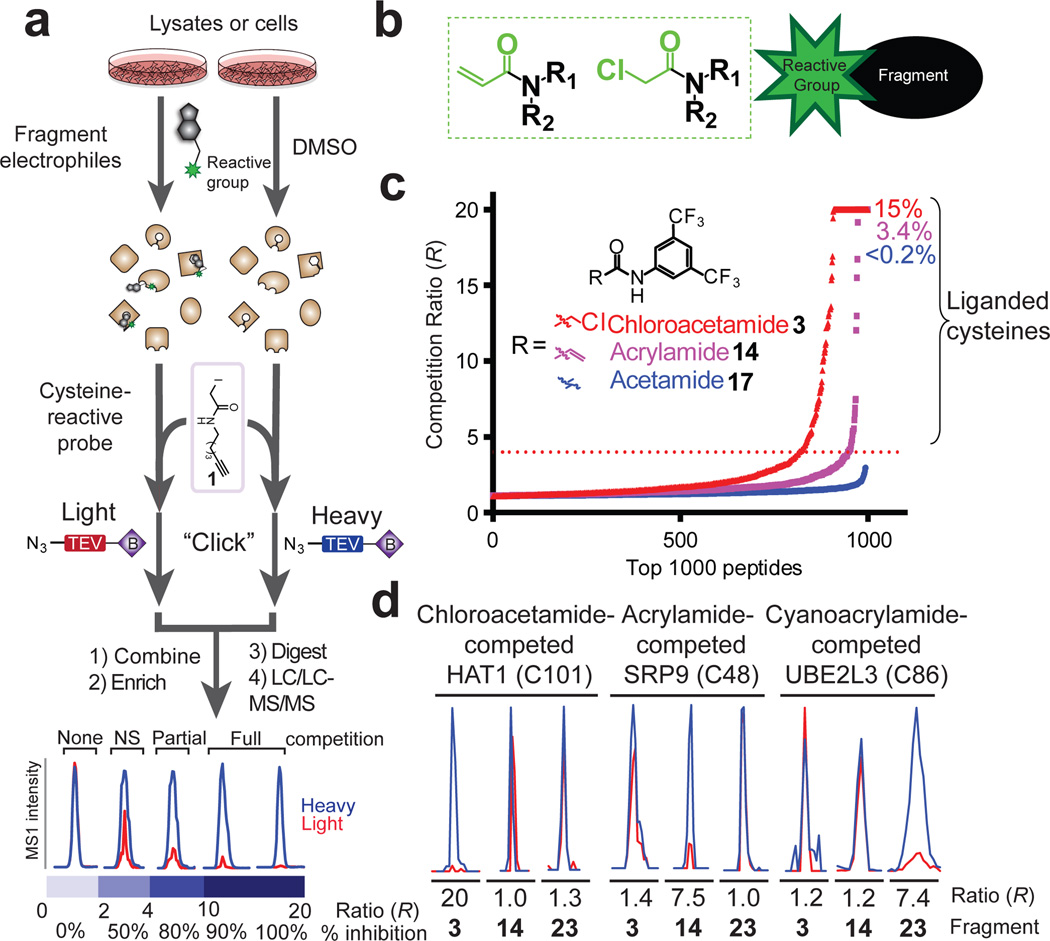
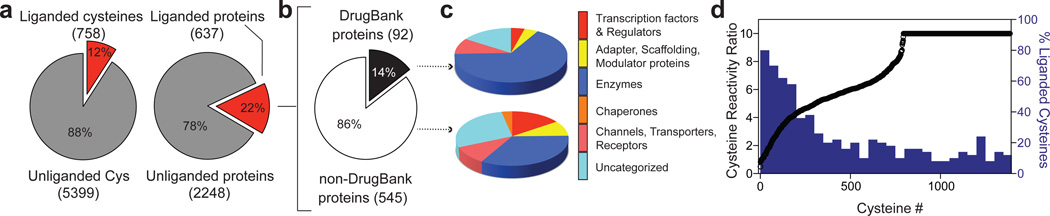
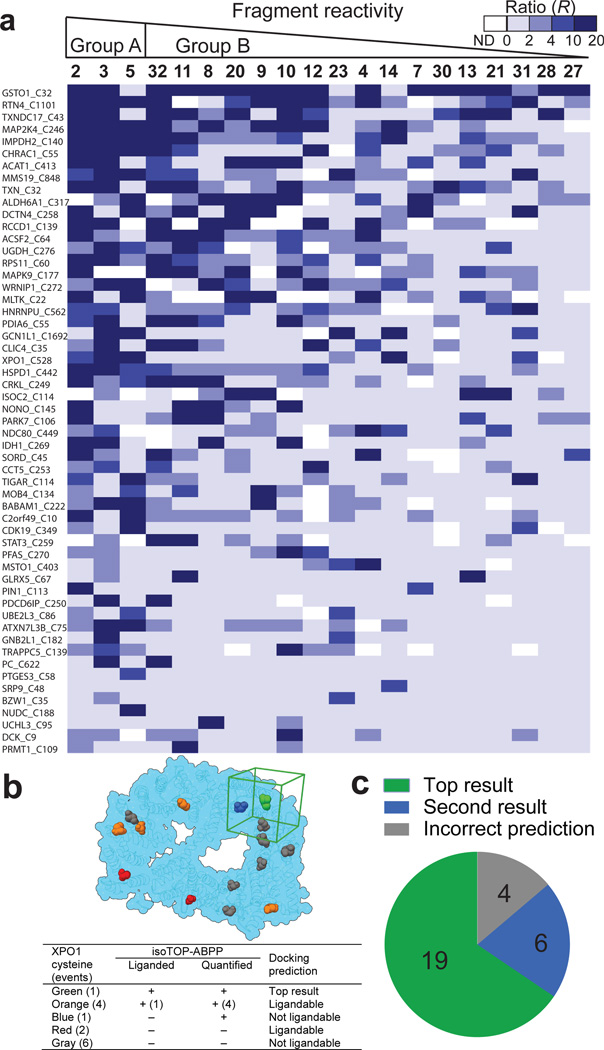
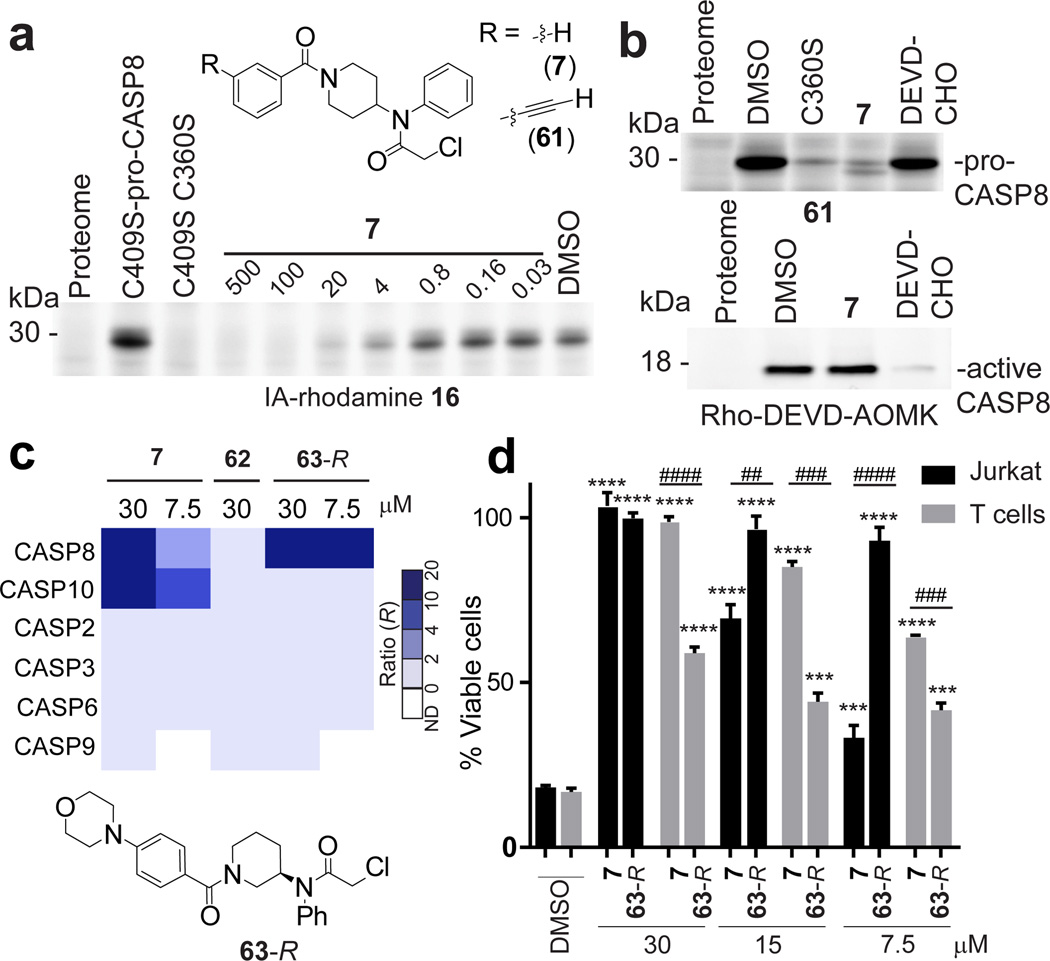
Small molecules are powerful tools for investigating protein function and can serve as leads for new therapeutics. Most human proteins, however, lack small-molecule ligands, and entire protein classes are considered 'undruggable'. Fragment-based ligand discovery can identify small-molecule probes for proteins that have proven difficult to target using high-throughput screening of complex compound libraries. Although reversibly binding ligands are commonly pursued, covalent fragments provide an alternative route to small-molecule probes, including those that can access regions of proteins that are difficult to target through binding affinity alone. Here we report a quantitative analysis of cysteine-reactive small-molecule fragments screened against thousands of proteins in human proteomes and cells. Covalent ligands were identified for >700 cysteines found in both druggable proteins and proteins deficient in chemical probes, including transcription factors, adaptor/scaffolding proteins, and uncharacterized proteins. Among the atypical ligand-protein interactions discovered were compounds that react preferentially with pro- (inactive) caspases. We used these ligands to distinguish extrinsic apoptosis pathways in human cell lines versus primary human T cells, showing that the former is largely mediated by caspase-8 while the latter depends on both caspase-8 and -10. Fragment-based covalent ligand discovery provides a greatly expanded portrait of the ligandable proteome and furnishes compounds that can illuminate protein functions in native biological systems.
Figures
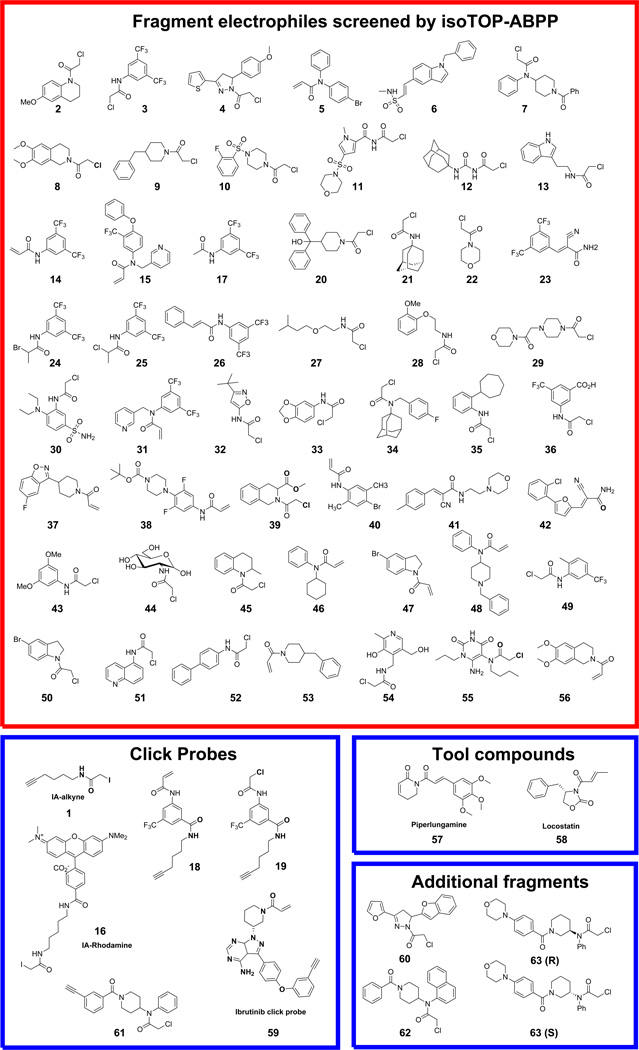
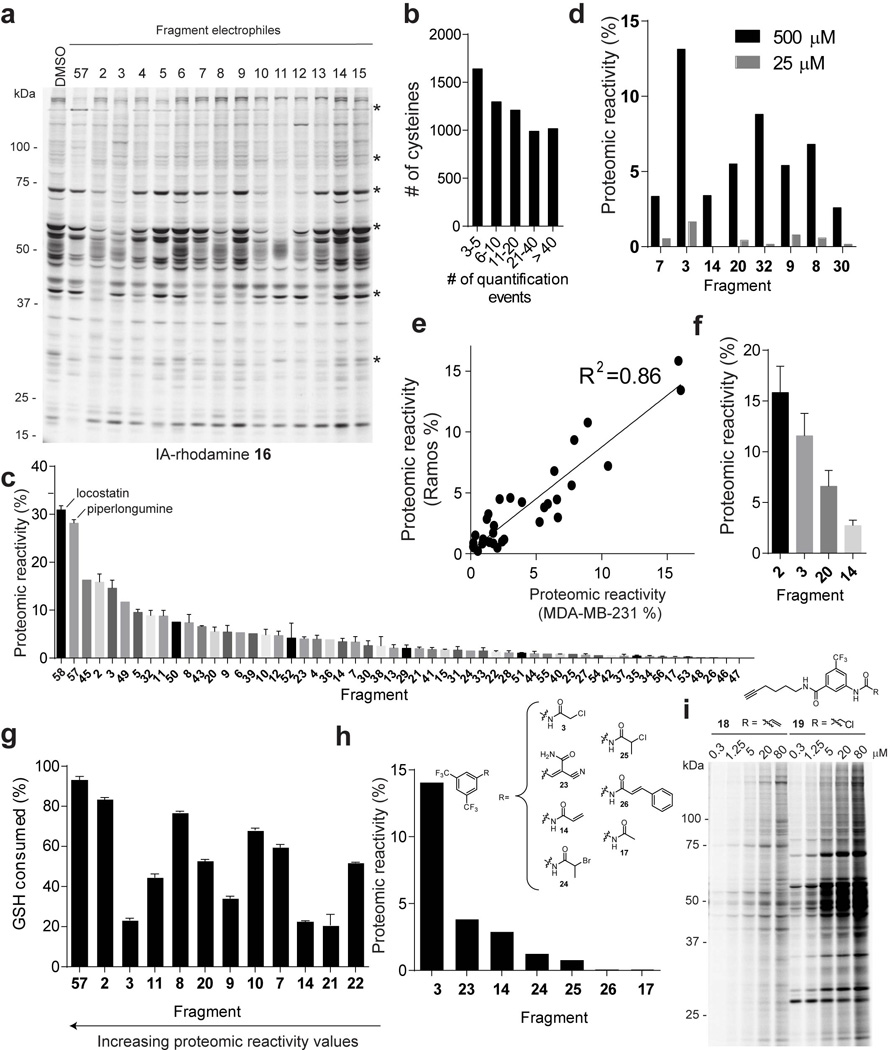
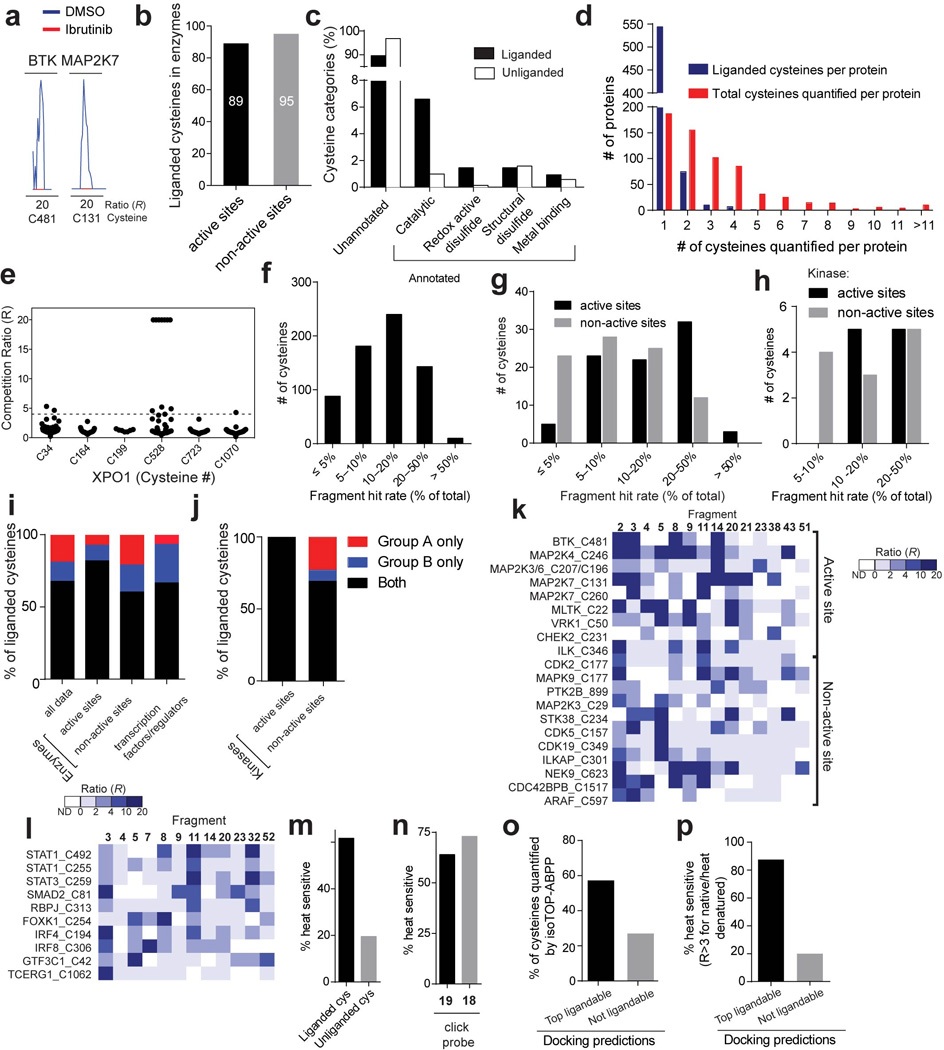
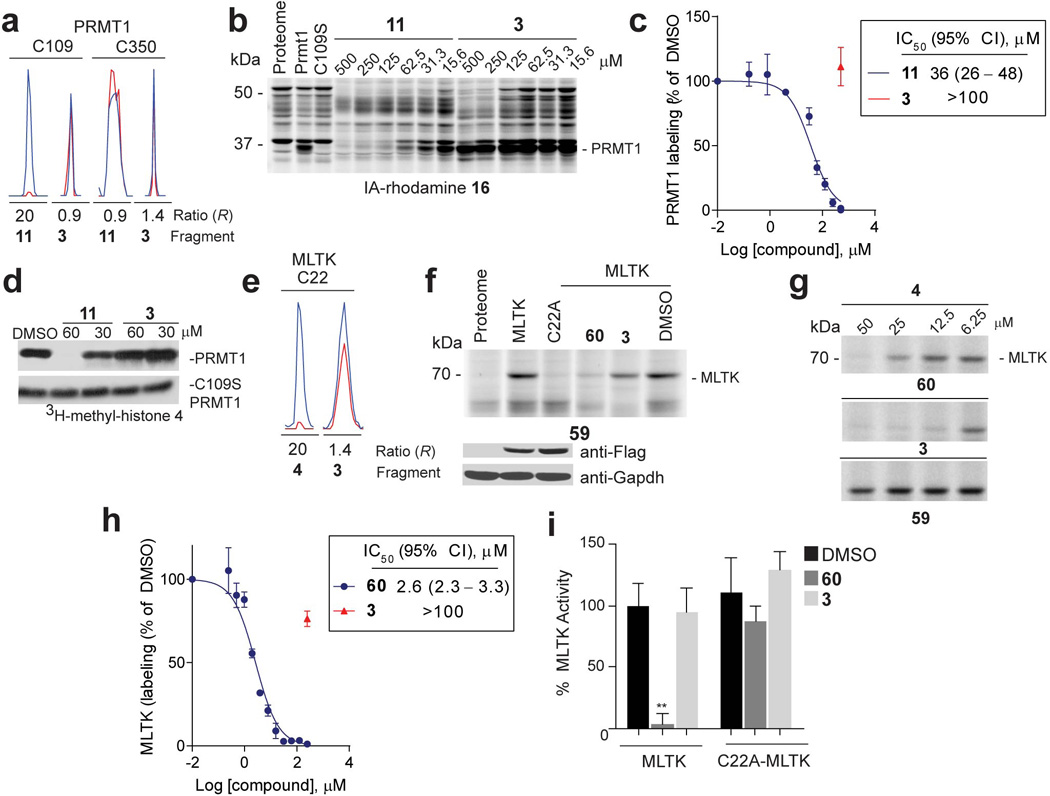
References
-
- Edfeldt FN, Folmer RH, Breeze AL. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug discovery today. 2011;16:284–287. - PubMed
-
- Hopkins AL, Groom CR. The druggable genome. Nature reviews. Drug discovery. 2002;1:727–730. - PubMed
-
- Scott DE, Coyne AG, Hudson SA, Abell C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry. 2012;51:4990–5003. - PubMed
References for online-only Methods
-
- Weerapana E, Speers AE, Cravatt BF. Tandem orthogonal proteolysis-activity-based protein profiling (TOP-ABPP)--a general method for mapping sites of probe modification in proteomes. Nat Protoc. 2007;2:1414–1425. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
