

Defining the consequences of genetic variation on a proteome-wide scale
- PMID: 27309819
- PMCID: PMC5292866
- DOI: 10.1038/nature18270
Defining the consequences of genetic variation on a proteome-wide scale
Erratum in
-
Author Correction: Defining the consequences of genetic variation on a proteome-wide scale.Nature. 2022 Jun;606(7915):E16. doi: 10.1038/s41586-022-04920-w. Nature. 2022. PMID: 35672473 No abstract available.
Abstract
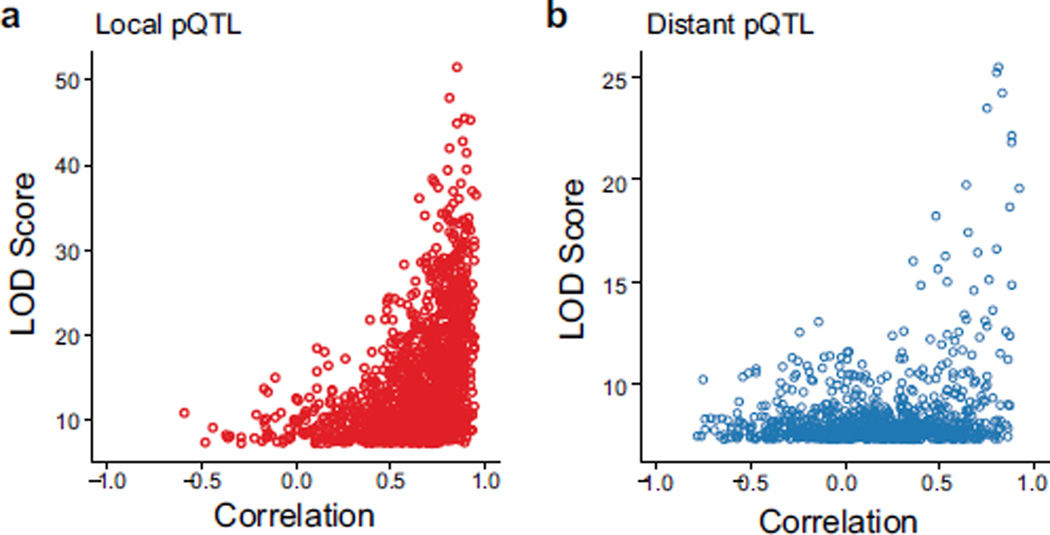
Genetic variation modulates protein expression through both transcriptional and post-transcriptional mechanisms. To characterize the consequences of natural genetic diversity on the proteome, here we combine a multiplexed, mass spectrometry-based method for protein quantification with an emerging outbred mouse model containing extensive genetic variation from eight inbred founder strains. By measuring genome-wide transcript and protein expression in livers from 192 Diversity outbred mice, we identify 2,866 protein quantitative trait loci (pQTL) with twice as many local as distant genetic variants. These data support distinct transcriptional and post-transcriptional models underlying the observed pQTL effects. Using a sensitive approach to mediation analysis, we often identified a second protein or transcript as the causal mediator of distant pQTL. Our analysis reveals an extensive network of direct protein-protein interactions. Finally, we show that local genotype can provide accurate predictions of protein abundance in an independent cohort of collaborative cross mice.
Figures














References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
