

Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism
- PMID: 27311541
- PMCID: PMC5028283
- DOI: 10.1016/j.neuint.2016.06.009
Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism
Abstract
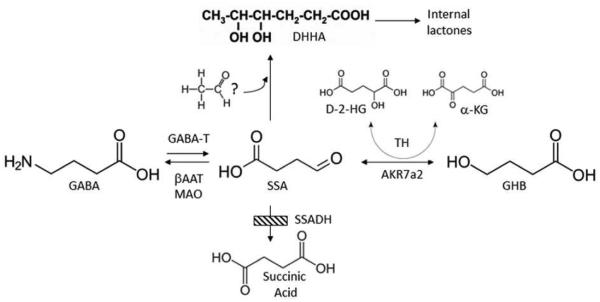
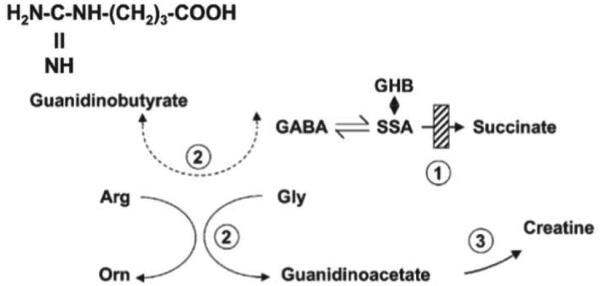
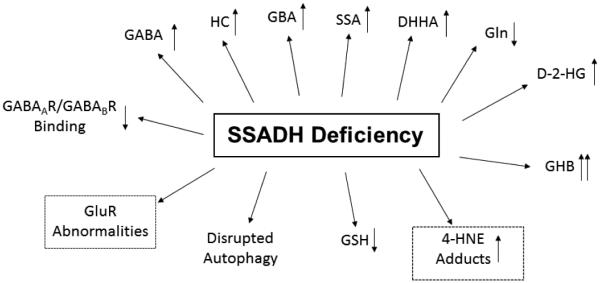
Discovered some 35 years ago, succinic semialdehyde dehydrogenase deficiency (SSADHD) represents a rare, autosomal recessively-inherited defect in the second step of the GABA degradative pathway. Some 200 patients have been reported, with broad phenotypic and genotypic heterogeneity. SSADHD represents an unusual neurometabolic disorder in which two neuromodulatory agents, GABA (and the GABA analogue, 4-hydroxybutyrate), accumulate to supraphysiological levels. The unexpected occurrence of epilepsy in several patients is counterintuitive in view of the hyperGABAergic state, in which sedation might be expected. However, the epileptic status of some patients is most likely represented by broader imbalances of GABAergic and glutamatergic neurotransmission. Cumulative research encompassing decades of basic and clinical study of SSADHD reveal a monogenic disease with broad pathophysiological and clinical phenotypes. Numerous metabolic perturbations unmasked in SSADHD include alterations in oxidative stress parameters, dysregulation of autophagy and mitophagy, dysregulation of both inhibitory and excitatory neurotransmitters and gene expression, and unique subsets of SNP alterations of the SSADH gene (so-called ALDH5A1, or aldehyde dehydrogenase 5A1 gene) on the 6p22 chromosomal arm. While seemingly difficult to collate and interpret, these anomalies have continued to open novel pathways for pharmacotherapeutic considerations. Here, we present an update on selected aspects of SSADHD, the ALDH5A1 gene, and future avenues for research on this rare disorder of GABA metabolism.
Keywords: Autophagy; Crystal structure; GABA (4-aminobutyric acid); GABAergic neurotransmission; GHB (4-hydroxybutyric acid); GWAS; Genome wide association study; Knockout mouse model; Mitophagy; Multifactorial traits; Neurological disease; Oxidative damage; Pathogenic mutations; Pathophysiology; Polymorphisms; SNP (single nucleotide polymorphism); Succinic semialdehyde dehydrogenase deficiency (SSADHD).
Copyright © 2016 Elsevier Ltd. All rights reserved.
Figures
References
-
- Al-Essa MA, Bakheet SM, Patay ZJ, Powe JE, Ozand PT. Clinical, fluorine-18 labeled 2-fluoro-2-deoxyglucose positron emission tomography (FDG PET) MRI of the brain and biochemical observations in a patient with 4-hydroxybutyric aciduria; a progressive neurometabolic disease. Brain Dev. 2000;22:127–31. - PubMed
-
- Ainslie GR, Gibson KM, Vogel KR. Maiese K, editor. mTOR, autophagy, aminoacidopathies, and human genetic disorders. Molecules to Medicine with mTOR, II. mTOR in Genetic Disorders and Neurodegenerative Disease. 2016:143–166. chap. 9.
-
- Akaboshi S, Hogema BM, Novelletto A, Malaspina P, Salomons GS, Maropoulos GD, Jakobs C, Grompe M, Gibson KM. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum. Mutat. 2003;22:442–450. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
