

Pseudotime estimation: deconfounding single cell time series
- PMID: 27318198
- PMCID: PMC5039927
- DOI: 10.1093/bioinformatics/btw372
Pseudotime estimation: deconfounding single cell time series
Abstract
Motivation: Repeated cross-sectional time series single cell data confound several sources of variation, with contributions from measurement noise, stochastic cell-to-cell variation and cell progression at different rates. Time series from single cell assays are particularly susceptible to confounding as the measurements are not averaged over populations of cells. When several genes are assayed in parallel these effects can be estimated and corrected for under certain smoothness assumptions on cell progression.
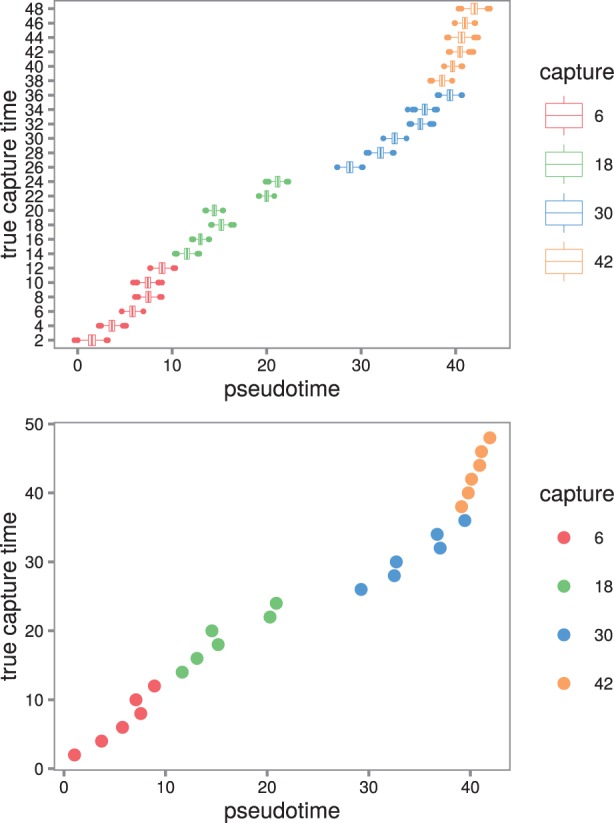
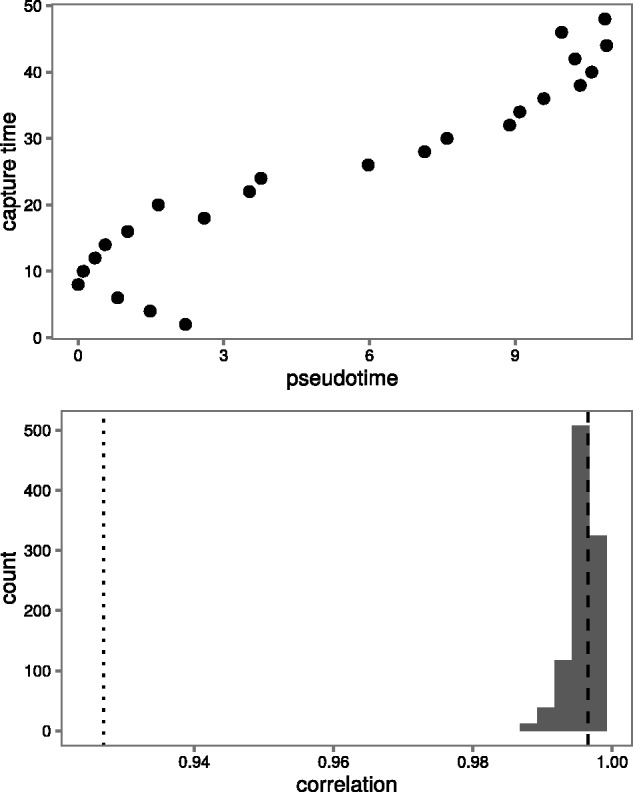
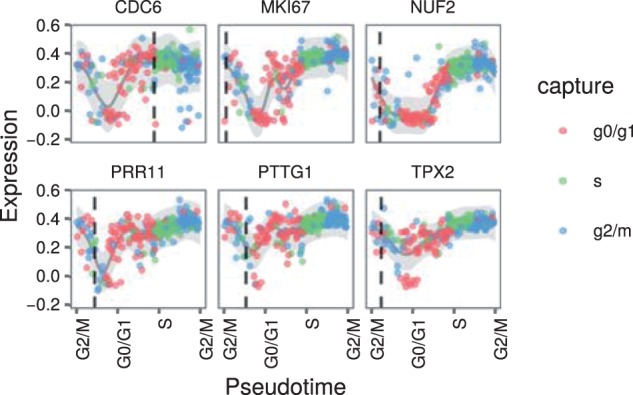
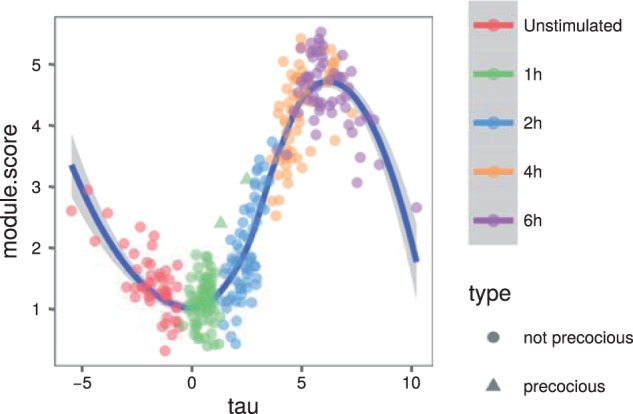
Results: We present a principled probabilistic model with a Bayesian inference scheme to analyse such data. We demonstrate our method's utility on public microarray, nCounter and RNA-seq datasets from three organisms. Our method almost perfectly recovers withheld capture times in an Arabidopsis dataset, it accurately estimates cell cycle peak times in a human prostate cancer cell line and it correctly identifies two precocious cells in a study of paracrine signalling in mouse dendritic cells. Furthermore, our method compares favourably with Monocle, a state-of-the-art technique. We also show using held-out data that uncertainty in the temporal dimension is a common confounder and should be accounted for in analyses of repeated cross-sectional time series.
Availability and implementation: Our method is available on CRAN in the DeLorean package.
Contact: john.reid@mrc-bsu.cam.ac.uk
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2016. Published by Oxford University Press.
Figures
References
-
- Äijö T., Lähdesmäki H. (2009) Learning gene regulatory networks from gene expression measurements using non-parametric molecular kinetics. Bioinformatics, 25, 2937–2944. - PubMed
-
- Brennecke P. et al. (2013) Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods, 10, 1093–1095. - PubMed
-
- Brooks S.P., Gelman A. (1998) General methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat., 7, 434–455.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
