

Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action?
- PMID: 27324312
- PMCID: PMC5023707
- DOI: 10.1113/JP270192
Arteriolar oxygen reactivity: where is the sensor and what is the mechanism of action?
Abstract
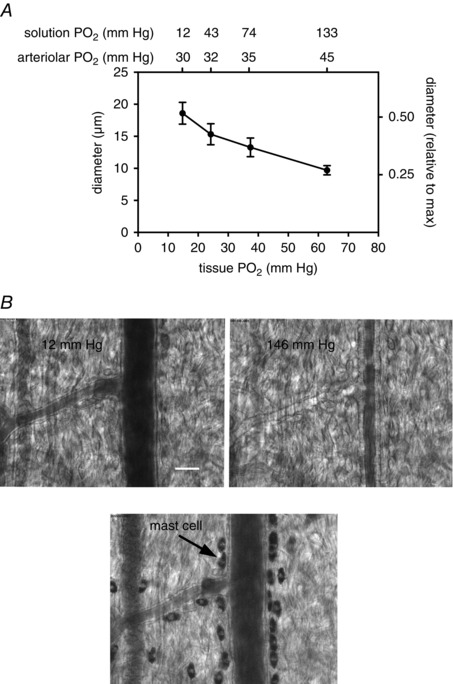
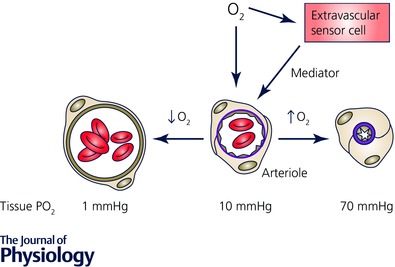
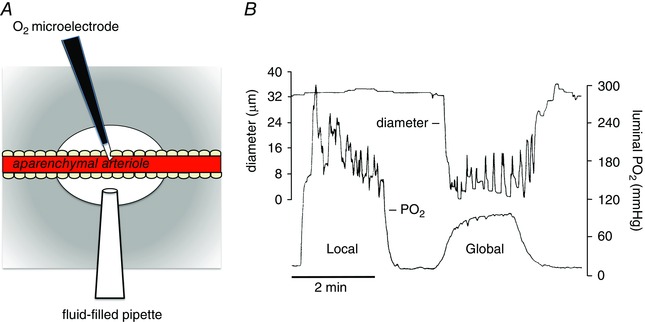
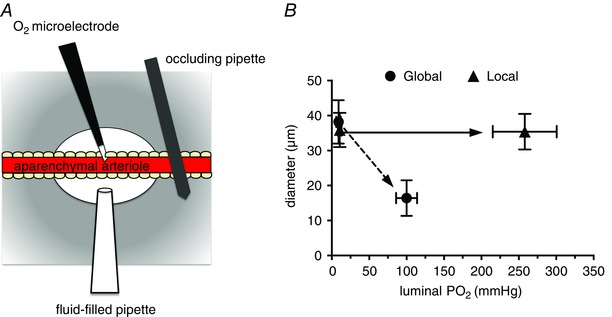
Arterioles in the peripheral microcirculation are exquisitely sensitive to changes in PO2 in their environment: increases in PO2 cause vasoconstriction while decreases in PO2 result in vasodilatation. However, the cell type that senses O2 (the O2 sensor) and the signalling pathway that couples changes in PO2 to changes in arteriolar tone (the mechanism of action) remain unclear. Many (but not all) ex vivo studies of isolated cannulated resistance arteries and large, first-order arterioles support the hypothesis that these vessels are intrinsically sensitive to PO2 with the smooth muscle, endothelial cells, or red blood cells serving as the O2 sensor. However, in situ studies testing these hypotheses in downstream arterioles have failed to find evidence of intrinsic O2 sensitivity, and instead have supported the idea that extravascular cells sense O2 . Similarly, ex vivo studies of isolated, cannulated resistance arteries and large first-order arterioles support the hypotheses that O2 -dependent inhibition of production of vasodilator cyclooxygenase products or O2 -dependent destruction of nitric oxide mediates O2 reactivity of these upstream vessels. In contrast, most in vivo studies of downstream arterioles have disproved these hypotheses and instead have provided evidence supporting the idea that O2 -dependent production of vasoconstrictors mediates arteriolar O2 reactivity, with significant regional heterogeneity in the specific vasoconstrictor involved. Oxygen-induced vasoconstriction may serve as a protective mechanism to reduce the oxidative burden to which a tissue is exposed, a process that is superimposed on top of the local mechanisms which regulate tissue blood flow to meet a tissue's metabolic demand.
Keywords: arterioles; microcirculation; oxygen; oxygen sensing; vasoconstriction; vasodilatation.
© 2016 The Authors. The Journal of Physiology © 2016 The Physiological Society.
Figures
Similar articles
-
The oxygen sensitivity of hamster cheek pouch arterioles. In vitro and in situ studies.Circ Res. 1983 Oct;53(4):515-25. doi: 10.1161/01.res.53.4.515. Circ Res. 1983. PMID: 6627610
-
Increases in oxygen tension evoke arteriolar constriction by inhibiting endothelial prostaglandin synthesis.Microvasc Res. 1994 Sep;48(2):151-60. doi: 10.1006/mvre.1994.1046. Microvasc Res. 1994. PMID: 7854203
-
Differential effect of cytochrome P-450 omega-hydroxylase inhibition on O2-induced constriction of arterioles in SHR with early and established hypertension.Microcirculation. 2001 Dec;8(6):435-43. doi: 10.1038/sj/mn/7800114. Microcirculation. 2001. PMID: 11781816
-
Molecular identification of O2 sensors and O2-sensitive potassium channels in the pulmonary circulation.Adv Exp Med Biol. 2000;475:219-40. doi: 10.1007/0-306-46825-5_21. Adv Exp Med Biol. 2000. PMID: 10849663 Review.
-
Regulation of coronary blood flow during exercise.Physiol Rev. 2008 Jul;88(3):1009-86. doi: 10.1152/physrev.00045.2006. Physiol Rev. 2008. PMID: 18626066 Review.
Cited by
-
Could respiration-driven blood oxygen changes modulate neural activity?Pflugers Arch. 2023 Jan;475(1):37-48. doi: 10.1007/s00424-022-02721-8. Epub 2022 Jun 28. Pflugers Arch. 2023. PMID: 35761104 Free PMC article. Review.
-
Sympathetic neurovascular transduction following acute hypoxia.Clin Auton Res. 2021 Dec;31(6):755-765. doi: 10.1007/s10286-021-00824-3. Epub 2021 Sep 15. Clin Auton Res. 2021. PMID: 34528146
-
Electro-metabolic signaling.J Gen Physiol. 2024 Feb 5;156(2):e202313451. doi: 10.1085/jgp.202313451. Epub 2024 Jan 10. J Gen Physiol. 2024. PMID: 38197953 Free PMC article.
-
Acute oxygen sensing by vascular smooth muscle cells.Front Physiol. 2023 Mar 3;14:1142354. doi: 10.3389/fphys.2023.1142354. eCollection 2023. Front Physiol. 2023. PMID: 36935756 Free PMC article. Review.
-
Hyperinsulinemia does not cause de novo capillary recruitment in rat skeletal muscle.Microcirculation. 2020 Feb;27(2):e12593. doi: 10.1111/micc.12593. Epub 2019 Oct 12. Microcirculation. 2020. PMID: 31605649 Free PMC article.
References
-
- Abu‐Soud HM, Ichimori K, Presta A & Stuehr DJ (2000). Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric‐oxide synthase. J Biol Chem 275, 17349–17357. - PubMed
-
- Baez S (1973). An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 5, 384–394. - PubMed
-
- Baron JF, Vicaut E, Hou X & Duvelleroy M (1990). Independent role of arterial O2 tension in local control of coronary blood flow. Am J Physiol 258, H1388–H1394. - PubMed
-
- Bartlett IS & Segal SS (2000). Resolution of smooth muscle and endothelial pathways for conduction along hamster cheek pouch arterioles. Am J Physiol Heart Circ Physiol 278, H604–H612. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources