

Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome
- PMID: 27325888
- PMCID: PMC4925017
- DOI: 10.1084/jem.20151529
Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome
Abstract
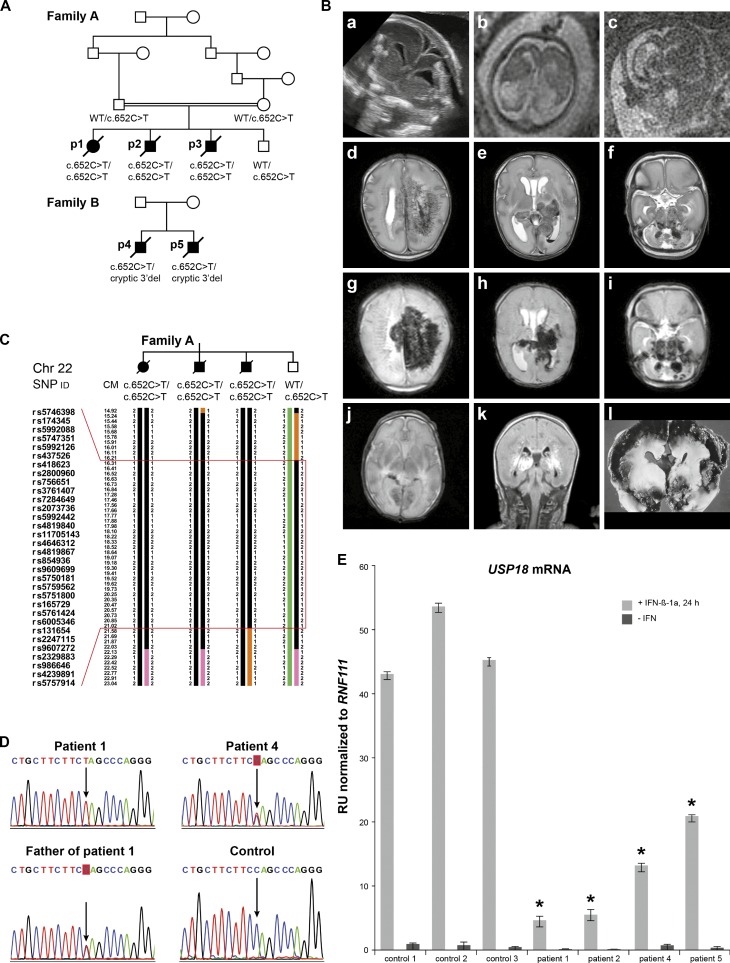
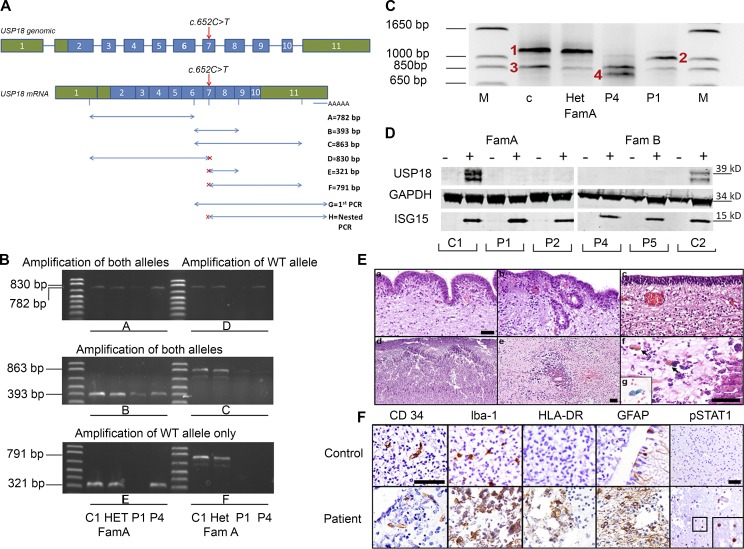
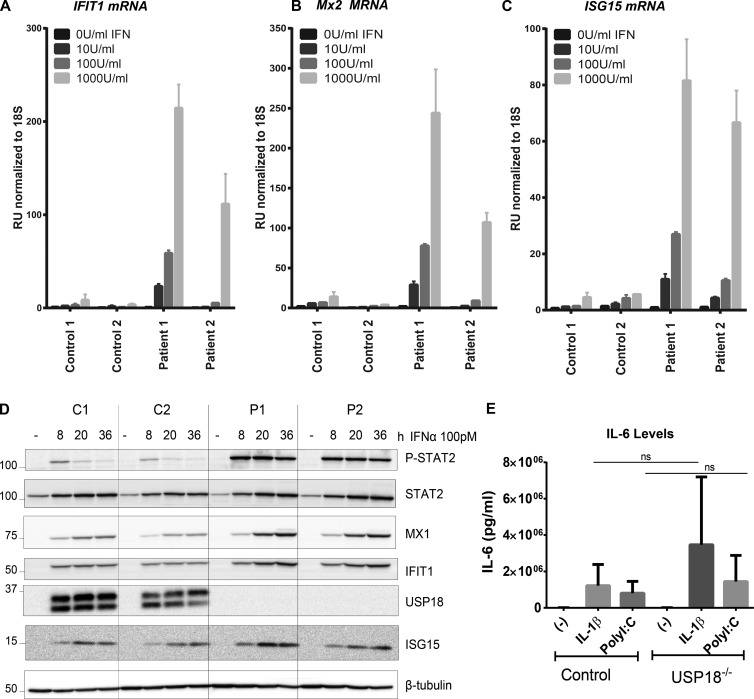
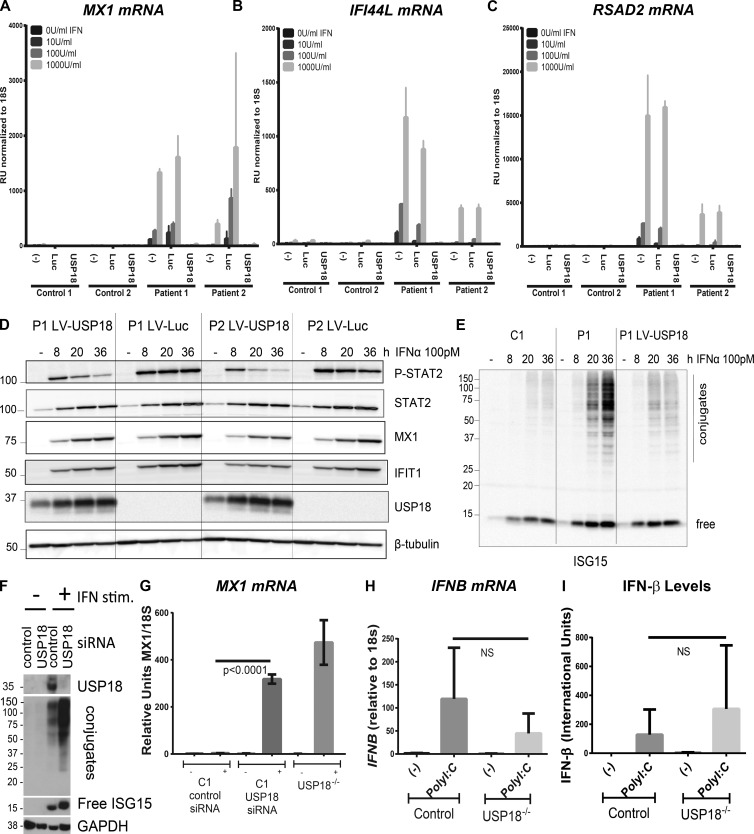
Pseudo-TORCH syndrome (PTS) is characterized by microcephaly, enlarged ventricles, cerebral calcification, and, occasionally, by systemic features at birth resembling the sequelae of congenital infection but in the absence of an infectious agent. Genetic defects resulting in activation of type 1 interferon (IFN) responses have been documented to cause Aicardi-Goutières syndrome, which is a cause of PTS. Ubiquitin-specific peptidase 18 (USP18) is a key negative regulator of type I IFN signaling. In this study, we identified loss-of-function recessive mutations of USP18 in five PTS patients from two unrelated families. Ex vivo brain autopsy material demonstrated innate immune inflammation with calcification and polymicrogyria. In vitro, patient fibroblasts displayed severely enhanced IFN-induced inflammation, which was completely rescued by lentiviral transduction of USP18. These findings add USP18 deficiency to the list of genetic disorders collectively termed type I interferonopathies. Moreover, USP18 deficiency represents the first genetic disorder of PTS caused by dysregulation of the response to type I IFNs. Therapeutically, this places USP18 as a promising target not only for genetic but also acquired IFN-mediated CNS disorders.
© 2016 Meuwissen et al.
Figures
References
-
- Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L., et al. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622. 10.1373/clinchem.2008.112797 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
