

Adjusting microbiome profiles for differences in microbial load by spike-in bacteria
- PMID: 27329048
- PMCID: PMC4915049
- DOI: 10.1186/s40168-016-0175-0
Adjusting microbiome profiles for differences in microbial load by spike-in bacteria
Abstract
Background: Next-generation 16S ribosomal RNA gene sequencing is widely used to determine the relative composition of the mammalian gut microbiomes. However, in the absence of a reference, this does not reveal alterations in absolute abundance of specific operational taxonomic units if microbial loads vary across specimens.
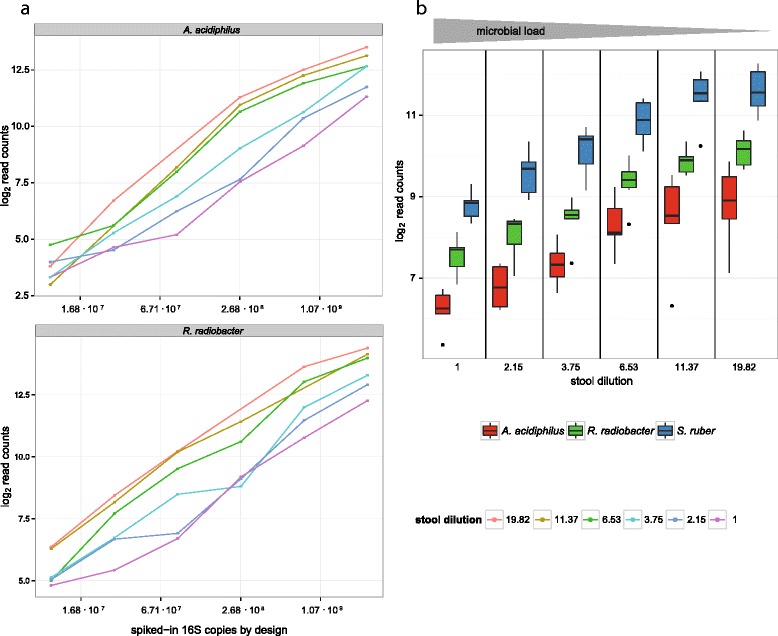
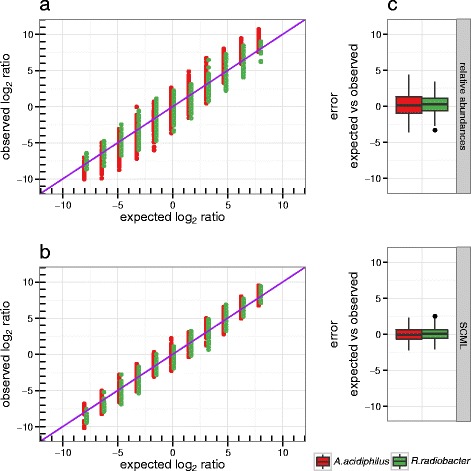
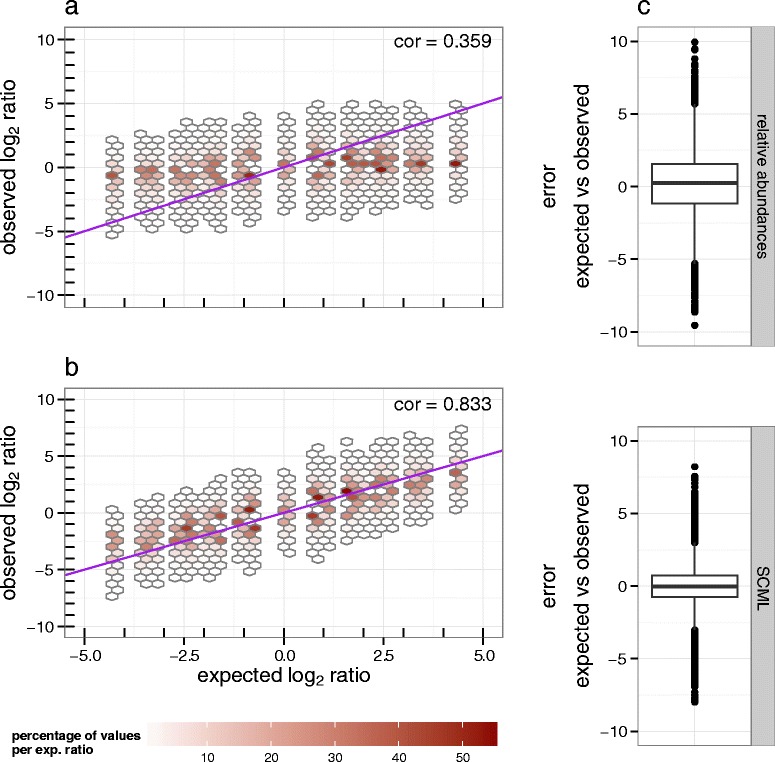
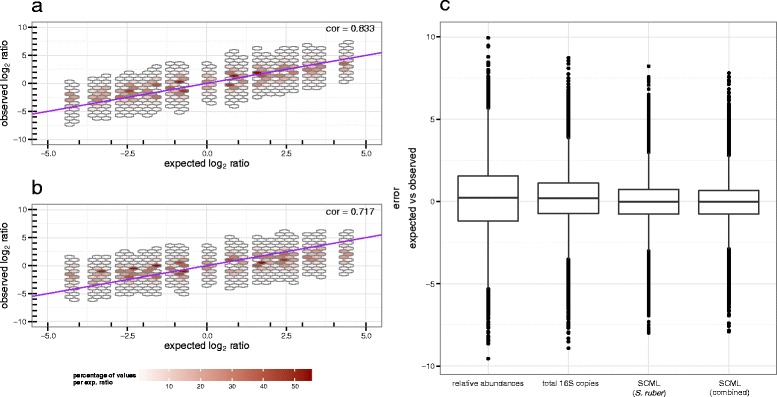
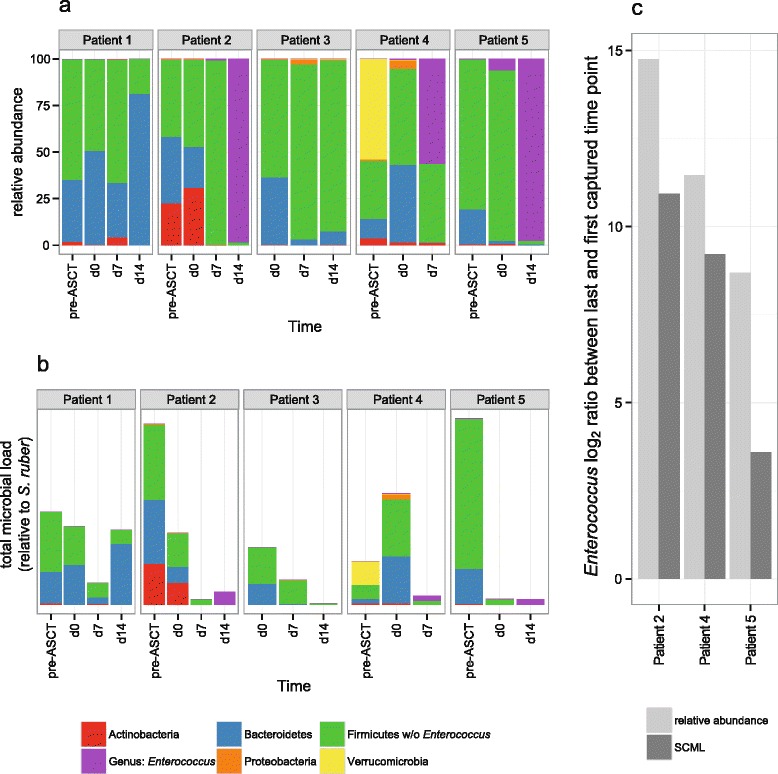
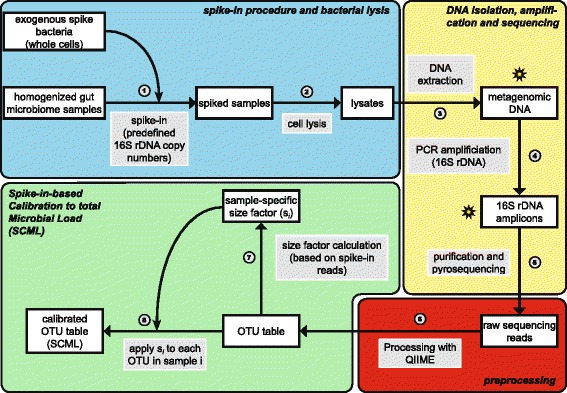
Results: Here we suggest the spiking of exogenous bacteria into crude specimens to quantify ratios of absolute bacterial abundances. We use the 16S rDNA read counts of the spike-in bacteria to adjust the read counts of endogenous bacteria for changes in total microbial loads. Using a series of dilutions of pooled faecal samples from mice containing defined amounts of the spike-in bacteria Salinibacter ruber, Rhizobium radiobacter and Alicyclobacillus acidiphilus, we demonstrate that spike-in-based calibration to microbial loads allows accurate estimation of ratios of absolute endogenous bacteria abundances. Applied to stool specimens of patients undergoing allogeneic stem cell transplantation, we were able to determine changes in both relative and absolute abundances of various phyla, especially the genus Enterococcus, in response to antibiotic treatment and radio-chemotherapeutic conditioning.
Conclusion: Exogenous spike-in bacteria in gut microbiome studies enable estimation of ratios of absolute OTU abundances, providing novel insights into the structure and the dynamics of intestinal microbiomes.
Keywords: 16S rRNA gene sequencing; Bacterial communities; Community analysis; Microbial load; Microbiome profiling; Spike-in bacteria; Standardization.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
