

Three families with 'de novo' m.3243A > G mutation
- PMID: 27331024
- PMCID: PMC4900294
- DOI: 10.1016/j.bbacli.2016.04.007
Three families with 'de novo' m.3243A > G mutation
Abstract
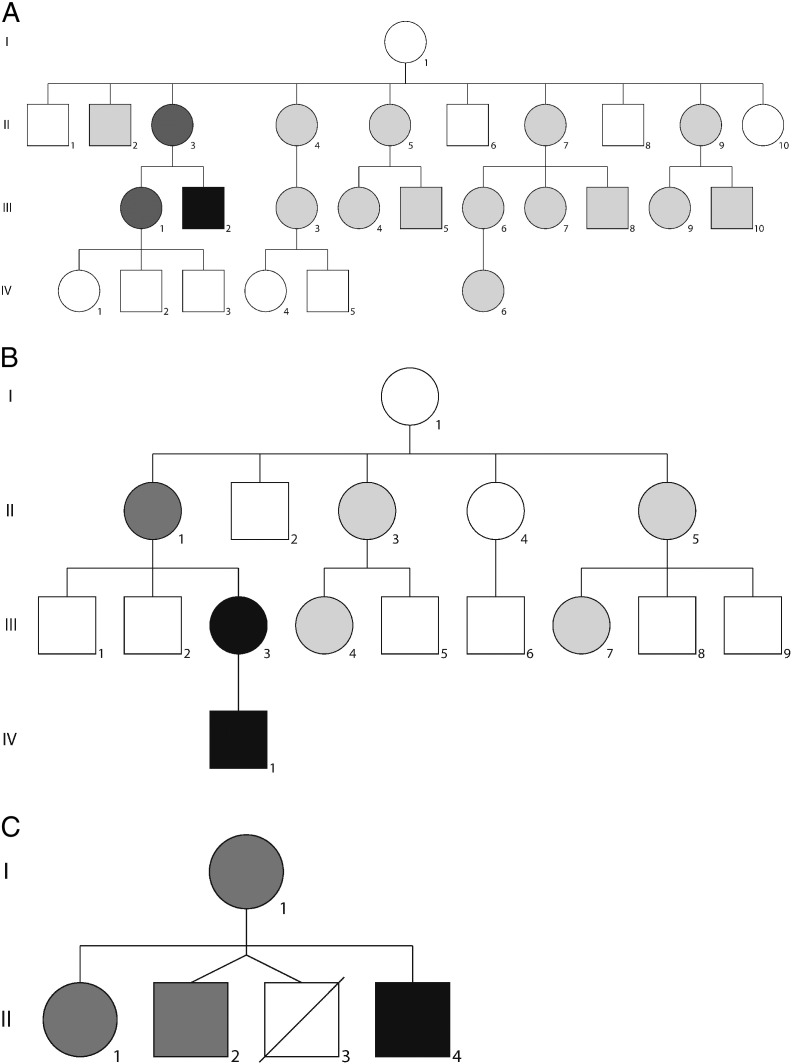
The m.3243A > G mutation is the most prevalent, disease-causing mitochondrial DNA (mtDNA) mutation. In a national cohort study of 48 families harbouring the m.3243A > G mutation, we identified three families in which the mutation appeared to occur sporadically within these families. In this report we describe these three families. Based on detailed mtDNA analysis of three different tissues using two different quantitative pyrosequencing assays with sensitivity to a level of 1% mutated mtDNA, we conclude that the m.3243A > G mutation has arisen de novo in each of these families. The symptomatic carriers presented with a variety of symptoms frequently observed in patients harbouring the m.3243A > G mutation. A more severe phenotype is seen in the de novo families compared to recent cohort studies, which might be due to reporting bias. The observation that de novo m.3243A > G mutations exist is of relevance for both diagnostic investigations and genetic counselling. Firstly, even where there is no significant (maternal) family history in patients with stroke-like episodes, diabetes and deafness or other unexplained organ dysfunction, the m.3243A > G mutation should be screened as a possible cause of the disease. Second, analysis of maternally-related family members is highly recommended to provide reliable counselling for these families, given that the m.3243A > G mutation may have arisen de novo.
Keywords: Genetic counselling; Inheritance; MELAS, mitochondrial myopathy, encephalopathy, lactate acidosis and stroke-like episodes; MERRF, myoclonic epilepsy with ragged-red fibres; MIDD, maternally inherited diabetes and deafness; Maternally inherited diabetes and deafness (MIDD); Mitochondrial myopathy, encephalopathy, lactate acidosis and stroke-like episodes (MELAS); m.3243A > G mutation; mtDNA, mitochondrial DNA.
Figures
Similar articles
-
One mutation, three phenotypes: novel metabolic insights on MELAS, MIDD and myopathy caused by the m.3243A > G mutation.Metabolomics. 2021 Jan 12;17(1):10. doi: 10.1007/s11306-020-01769-w. Metabolomics. 2021. PMID: 33438095
-
De Novo Mutation of m.3243A>G together with m.16093T>C Associated with Atypical Clinical Features in a Pedigree with MIDD Syndrome.J Diabetes Res. 2019 Apr 4;2019:5184647. doi: 10.1155/2019/5184647. eCollection 2019. J Diabetes Res. 2019. PMID: 31143779 Free PMC article.
-
The heart in m.3243A>G carriers.Herz. 2020 Jun;45(4):356-361. doi: 10.1007/s00059-018-4739-6. Epub 2018 Aug 20. Herz. 2020. PMID: 30128910 Review. English.
-
The UK MRC Mitochondrial Disease Patient Cohort Study: clinical phenotypes associated with the m.3243A>G mutation--implications for diagnosis and management.J Neurol Neurosurg Psychiatry. 2013 Aug;84(8):936-8. doi: 10.1136/jnnp-2012-303528. Epub 2013 Jan 25. J Neurol Neurosurg Psychiatry. 2013. PMID: 23355809
-
Renal manifestations in adults with mitochondrial disease from the mtDNA m.3243A>G pathogenic variant.Nefrologia (Engl Ed). 2023 Dec;43 Suppl 2:1-7. doi: 10.1016/j.nefroe.2024.01.017. Nefrologia (Engl Ed). 2023. PMID: 38355238 Review.
Cited by
-
Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors.Ann Clin Transl Neurol. 2018 Feb 7;5(3):333-345. doi: 10.1002/acn3.532. eCollection 2018 Mar. Ann Clin Transl Neurol. 2018. PMID: 29560378 Free PMC article.
-
Ancient mitochondrial DNA pathogenic variants putatively associated with mitochondrial disease.PLoS One. 2020 Sep 24;15(9):e0233666. doi: 10.1371/journal.pone.0233666. eCollection 2020. PLoS One. 2020. PMID: 32970680 Free PMC article.
-
T cell differentiation drives the negative selection of pathogenic mitochondrial DNA variants.Life Sci Alliance. 2023 Aug 31;6(11):e202302271. doi: 10.26508/lsa.202302271. Print 2023 Nov. Life Sci Alliance. 2023. PMID: 37652671 Free PMC article.
-
mtDNA heteroplasmy level and copy number indicate disease burden in m.3243A>G mitochondrial disease.EMBO Mol Med. 2018 Jun;10(6):e8262. doi: 10.15252/emmm.201708262. EMBO Mol Med. 2018. PMID: 29735722 Free PMC article.
-
Genetic Counselling for Maternally Inherited Mitochondrial Disorders.Mol Diagn Ther. 2017 Aug;21(4):419-429. doi: 10.1007/s40291-017-0279-7. Mol Diagn Ther. 2017. PMID: 28536827 Review.
References
-
- Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. - PubMed
-
- Schwartz M., Vissing J. No evidence for paternal inheritance of mtDNA in patients with sporadic mtDNA mutations. J. Neurol. Sci. 2004;218:99–101. - PubMed
-
- Chinnery P.F., DiMauro S., Shanske S., Schon E.A., Zeviani M., Mariotti C., Carrara F., Lombes A., Laforet P., Ogier H., Jaksch M., Lochmuller H., Horvath R., Deschauer M., Thorburn D.R., Bindoff L.A., Poulton J., Taylor R.W., Matthews J.N., Turnbull D.M. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004;364:592–596. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
