

TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3
- PMID: 27333864
- PMCID: PMC4917863
- DOI: 10.1038/srep28299
TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3
Abstract
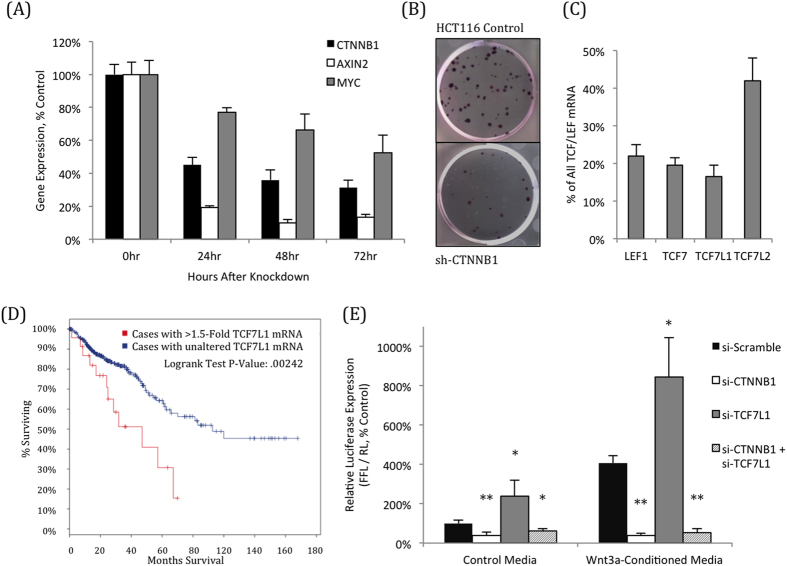
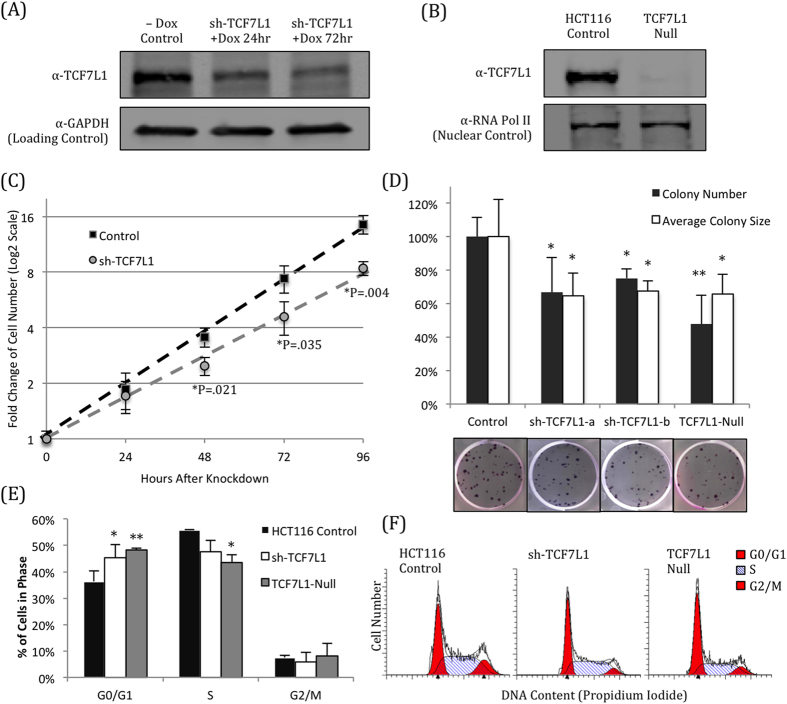
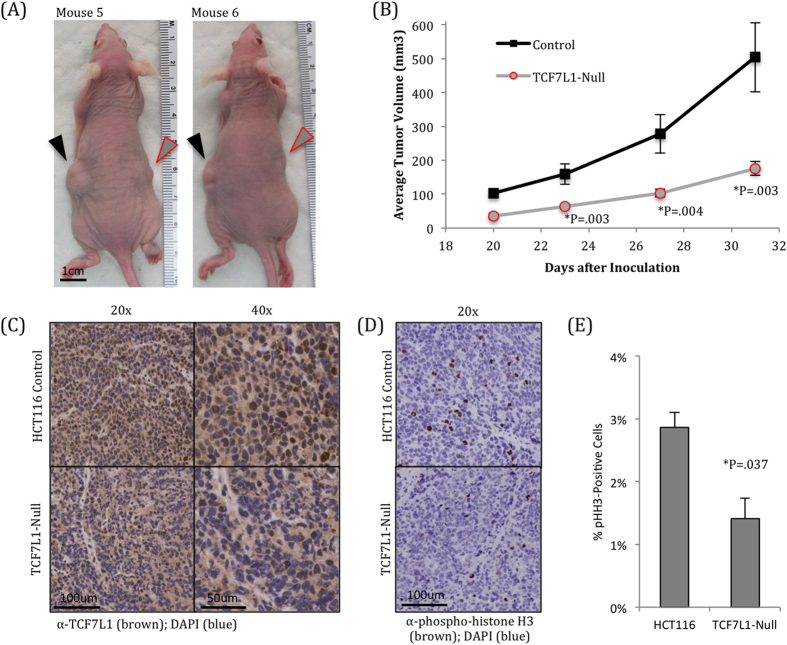
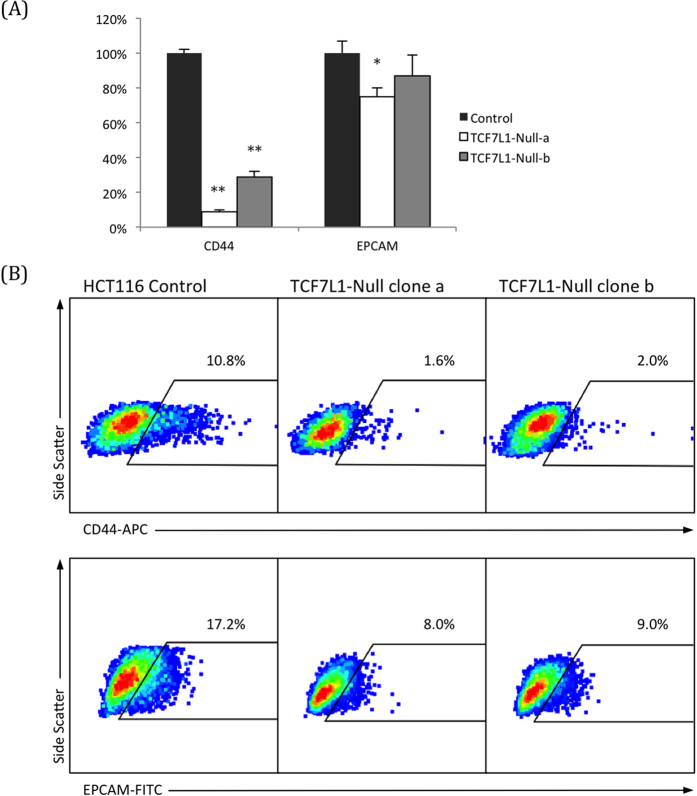
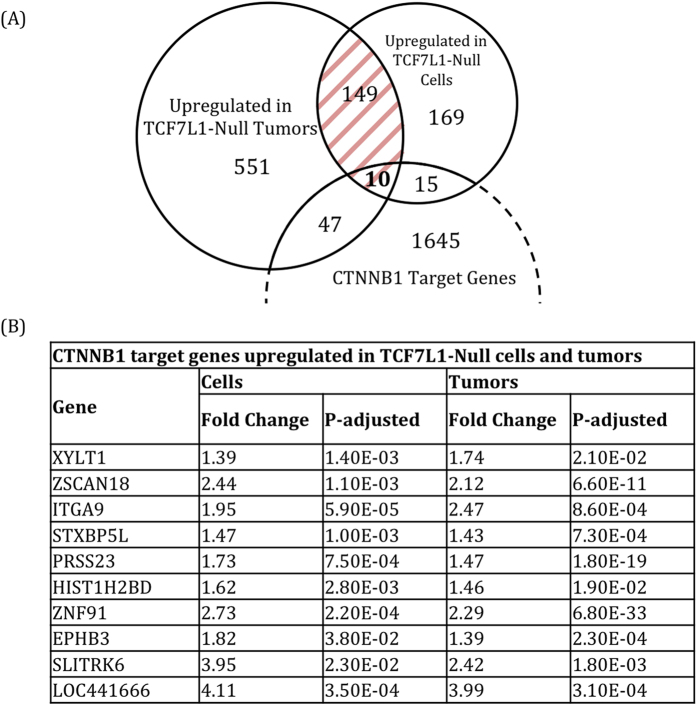
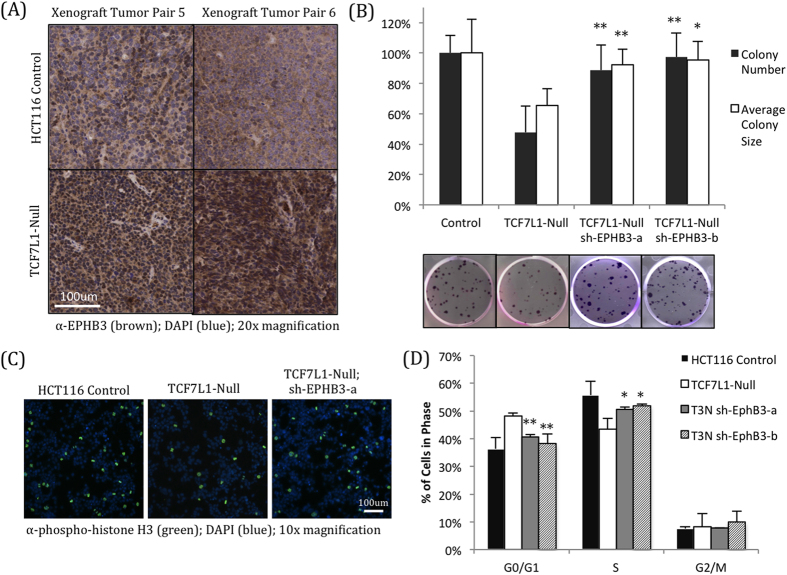
Dysregulation of the Wnt pathway leading to accumulation of β-catenin (CTNNB1) is a hallmark of colorectal cancer (CRC). Nuclear CTNNB1 acts as a transcriptional coactivator with TCF/LEF transcription factors, promoting expression of a broad set of target genes, some of which promote tumor growth. However, it remains poorly understood how CTNNB1 interacts with different transcription factors in different contexts to promote different outcomes. While some CTNNB1 target genes are oncogenic, others regulate differentiation. Here, we found that TCF7L1, a Wnt pathway repressor, buffers CTNNB1/TCF target gene expression to promote CRC growth. Loss of TCF7L1 impaired growth and colony formation of HCT116 CRC cells and reduced tumor growth in a mouse xenograft model. We identified a group of CTNNB1/TCF target genes that are activated in the absence of TCF7L1, including EPHB3, a marker of Paneth cell differentiation that has also been implicated as a tumor suppressor in CRC. Knockdown of EPHB3 partially restores growth and normal cell cycle progression of TCF7L1-Null cells. These findings suggest that while CTNNB1 accumulation is critical for CRC progression, activation of specific Wnt target genes in certain contexts may in fact inhibit tumor growth.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
