

Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses
- PMID: 27334580
- PMCID: PMC4988138
- DOI: 10.1128/JVI.00832-16
Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses
Abstract
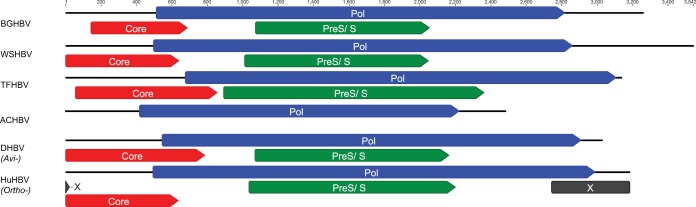
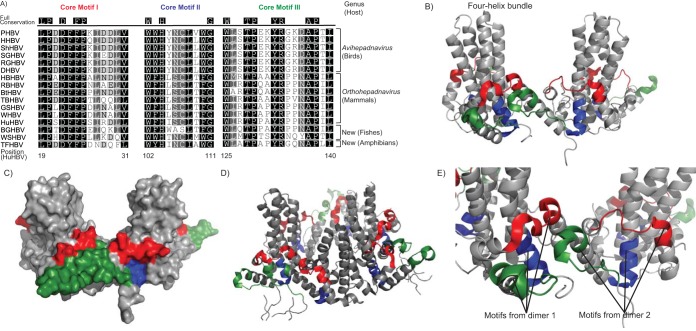
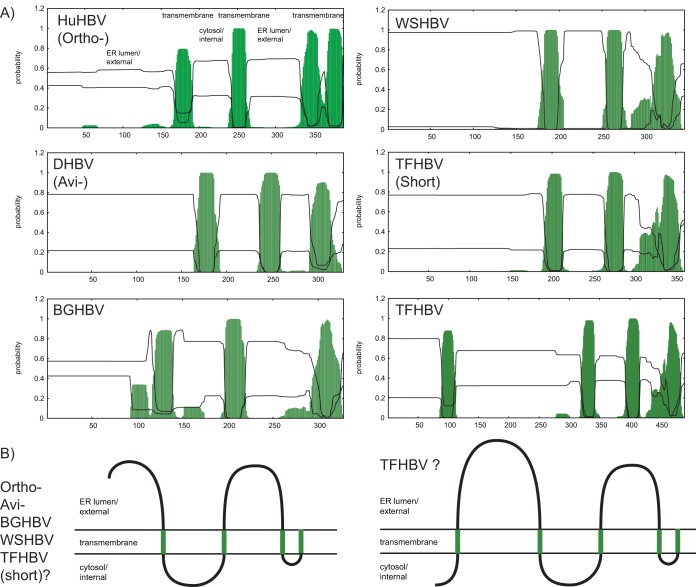
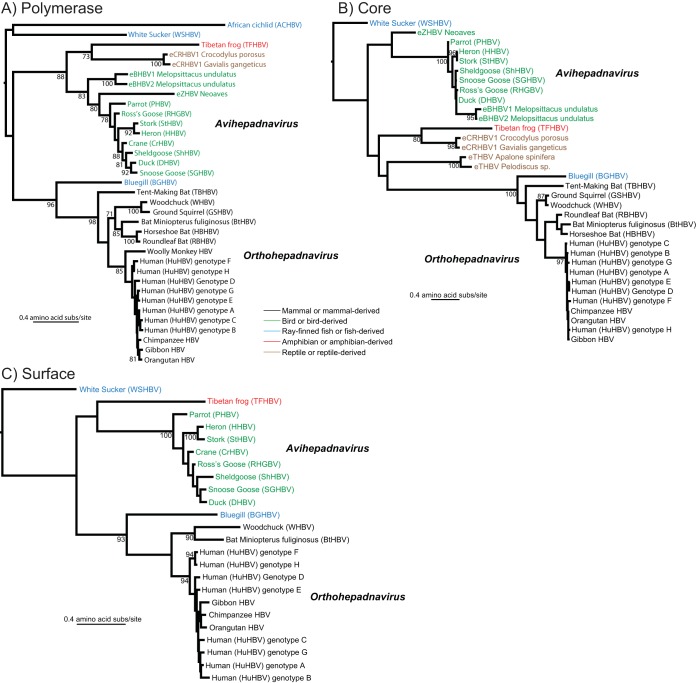
Hepadnaviruses (hepatitis B viruses [HBVs]) are the only animal viruses that replicate their DNA by reverse transcription of an RNA intermediate. Until recently, the known host range of hepadnaviruses was limited to mammals and birds. We obtained and analyzed the first amphibian HBV genome, as well as several prototype fish HBVs, which allow the first comprehensive comparative genomic analysis of hepadnaviruses from four classes of vertebrates. Bluegill hepadnavirus (BGHBV) was characterized from in-house viral metagenomic sequencing. The African cichlid hepadnavirus (ACHBV) and the Tibetan frog hepadnavirus (TFHBV) were discovered using in silico analyses of the whole-genome shotgun and transcriptome shotgun assembly databases. Residues in the hydrophobic base of the capsid (core) proteins, designated motifs I, II, and III, are highly conserved, suggesting that structural constraints for proper capsid folding are key to capsid protein evolution. Surface proteins in all vertebrate HBVs contain similar predicted membrane topologies, characterized by three transmembrane domains. Most striking was the fact that BGHBV, ACHBV, and the previously described white sucker hepadnavirus did not form a fish-specific monophyletic group in the phylogenetic analysis of all three hepadnaviral genes. Notably, BGHBV was more closely related to the mammalian hepadnaviruses, indicating that cross-species transmission events have played a major role in viral evolution. Evidence of cross-species transmission was also observed with TFHBV. Hence, these data indicate that the evolutionary history of the hepadnaviruses is more complex than previously realized and combines both virus-host codivergence over millions of years and host species jumping.
Importance: Hepadnaviruses are responsible for significant disease in humans (hepatitis B virus) and have been reported from a diverse range of vertebrates as both exogenous and endogenous viruses. We report the full-length genome of a novel hepadnavirus from a fish and the first hepadnavirus genome from an amphibian. The novel fish hepadnavirus, sampled from bluegills, was more closely related to mammalian hepadnaviruses than to other fish viruses. This phylogenetic pattern reveals that, although hepadnaviruses have likely been associated with vertebrates for hundreds of millions of years, they have also been characterized by species jumping across wide phylogenetic distances.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures


Similar articles
-
Characterization of a Novel Hepadnavirus in the White Sucker (Catostomus commersonii) from the Great Lakes Region of the United States.J Virol. 2015 Dec;89(23):11801-11. doi: 10.1128/JVI.01278-15. Epub 2015 Sep 16. J Virol. 2015. PMID: 26378165 Free PMC article.
-
Complete genome sequence of a divergent strain of Tibetan frog hepatitis B virus associated with a concave-eared torrent frog (Odorrana tormota).Arch Virol. 2019 Jun;164(6):1727-1732. doi: 10.1007/s00705-019-04227-8. Epub 2019 Mar 28. Arch Virol. 2019. PMID: 30923967
-
Early mesozoic coexistence of amniotes and hepadnaviridae.PLoS Genet. 2014 Dec 11;10(12):e1004559. doi: 10.1371/journal.pgen.1004559. eCollection 2014 Dec. PLoS Genet. 2014. PMID: 25501991 Free PMC article.
-
Tracing the evolutionary history of hepadnaviruses in terms of e antigen and middle envelope protein expression or processing.Virus Res. 2020 Jan 15;276:197825. doi: 10.1016/j.virusres.2019.197825. Epub 2019 Nov 27. Virus Res. 2020. PMID: 31785305 Free PMC article. Review.
-
Bat hepadnaviruses and the origins of primate hepatitis B viruses.Curr Opin Virol. 2016 Feb;16:86-94. doi: 10.1016/j.coviro.2016.01.015. Epub 2016 Feb 18. Curr Opin Virol. 2016. PMID: 26897577 Review.
Cited by
-
Discovery of a highly divergent hepadnavirus in shrews from China.Virology. 2019 May;531:162-170. doi: 10.1016/j.virol.2019.03.007. Epub 2019 Mar 11. Virology. 2019. PMID: 30884426 Free PMC article.
-
HDV-Like Viruses.Viruses. 2021 Jun 23;13(7):1207. doi: 10.3390/v13071207. Viruses. 2021. PMID: 34201626 Free PMC article. Review.
-
Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses.Cell Host Microbe. 2017 Sep 13;22(3):387-399.e6. doi: 10.1016/j.chom.2017.07.019. Epub 2017 Aug 31. Cell Host Microbe. 2017. PMID: 28867387 Free PMC article.
-
Novel Avulaviruses in Penguins, Antarctica.Emerg Infect Dis. 2017 Jul;23(7):1212-1214. doi: 10.3201/eid2307.170054. Emerg Infect Dis. 2017. PMID: 28628443 Free PMC article.
-
Metagenomic analysis uncovers novel hepadnaviruses and nackednaviruses.Sci Rep. 2025 Jul 9;15(1):24699. doi: 10.1038/s41598-025-05993-z. Sci Rep. 2025. PMID: 40634364 Free PMC article.
References
-
- Seeger C, Zoulim F, Mason WS. 2013. Hepadnaviruses, p 2185–2221. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA.
-
- Voyles BA. 1993. The biology of viruses. Mosby, St. Louis, MO.
-
- Flint SJ, Enquist LW, Racaniello VR, Skalka AM, Barnum DR, de Evaluación E. 2000. Principles of virology: molecular biology, pathogenesis and control. ASM Press, Washington, DC.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
