

Opposing Roles of Double-Stranded RNA Effector Pathways and Viral Defense Proteins Revealed with CRISPR-Cas9 Knockout Cell Lines and Vaccinia Virus Mutants
- PMID: 27334583
- PMCID: PMC4988158
- DOI: 10.1128/JVI.00869-16
Opposing Roles of Double-Stranded RNA Effector Pathways and Viral Defense Proteins Revealed with CRISPR-Cas9 Knockout Cell Lines and Vaccinia Virus Mutants
Abstract
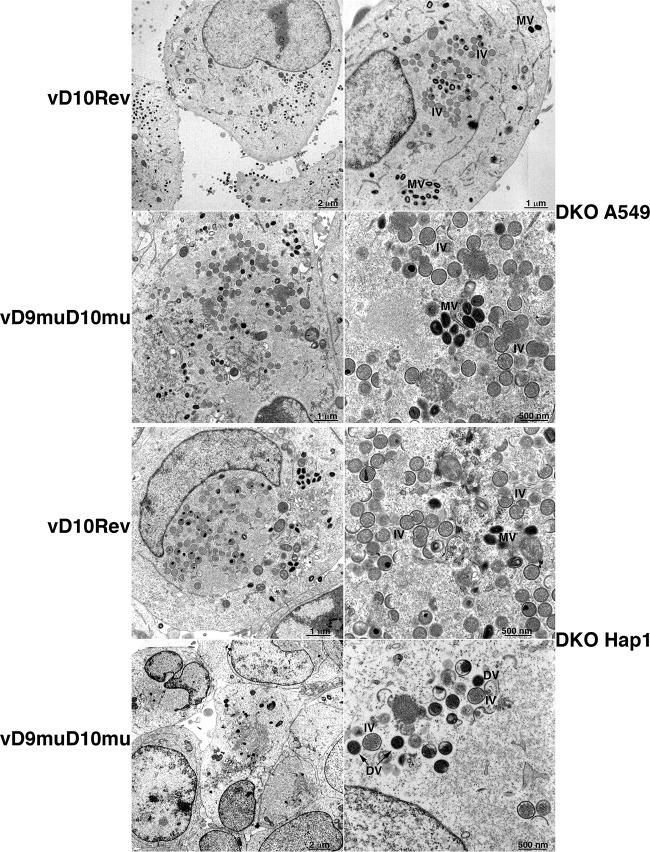
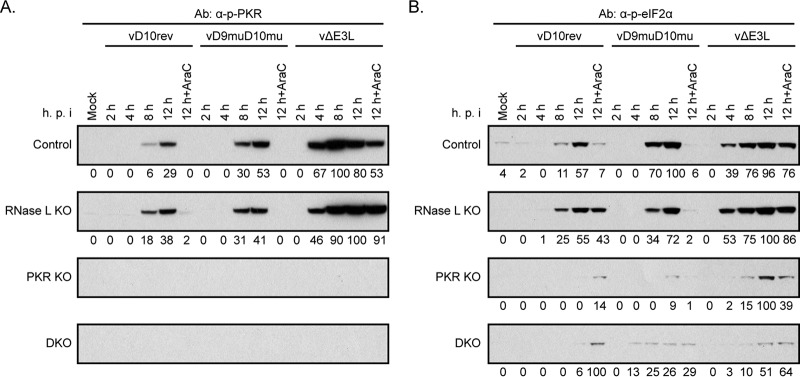
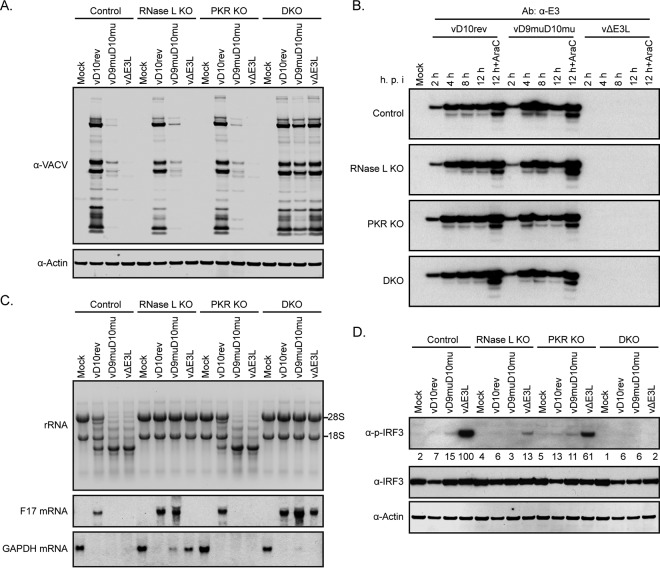
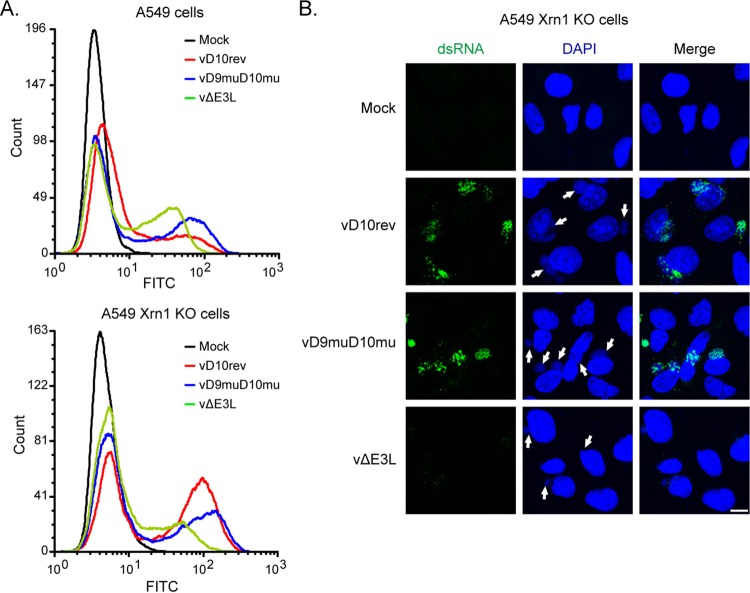
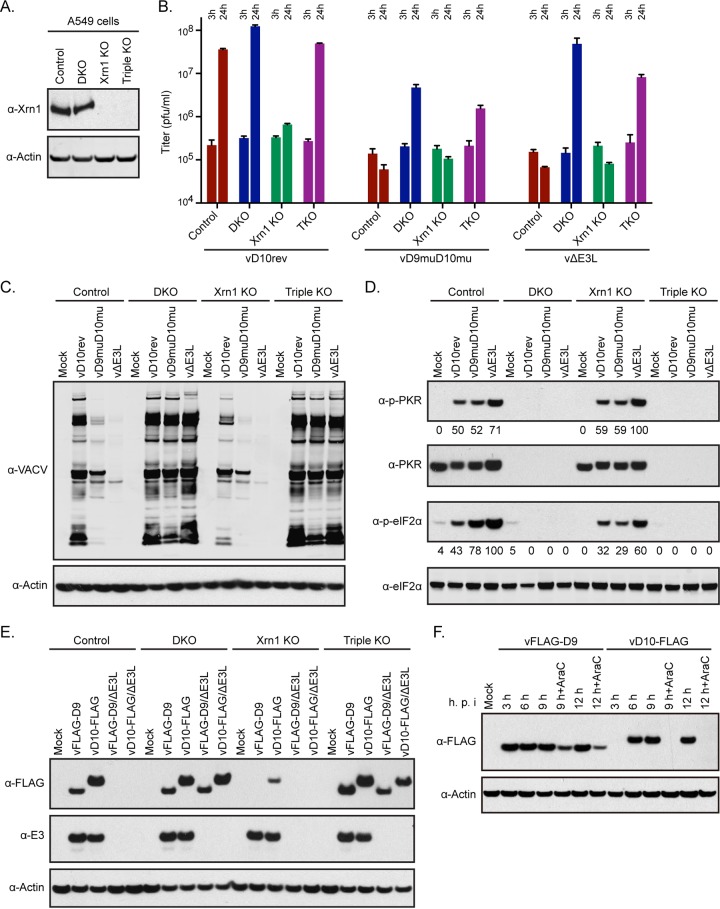
Vaccinia virus (VACV) decapping enzymes and cellular exoribonuclease Xrn1 catalyze successive steps in mRNA degradation and prevent double-stranded RNA (dsRNA) accumulation, whereas the viral E3 protein can bind dsRNA. We showed that dsRNA and E3 colocalized within cytoplasmic viral factories in cells infected with a decapping enzyme mutant as well as with wild-type VACV and that they coprecipitated with antibody. An E3 deletion mutant induced protein kinase R (PKR) and eukaryotic translation initiation factor alpha (eIF2α) phosphorylation earlier and more strongly than a decapping enzyme mutant even though less dsRNA was made, leading to more profound effects on viral gene expression. Human HAP1 and A549 cells were genetically modified by clustered regularly interspaced short palindromic repeat-Cas9 (CRISPR-Cas9) to determine whether the same pathways restrict E3 and decapping mutants. The E3 mutant replicated in PKR knockout (KO) HAP1 cells in which RNase L is intrinsically inactive but only with a double knockout (DKO) of PKR and RNase L in A549 cells, indicating that both pathways decreased replication equivalently and that no additional dsRNA pathway was crucial. In contrast, replication of the decapping enzyme mutant increased significantly (though less than that of wild-type virus) in DKO A549 cells but not in DKO HAP1 cells where a smaller increase in viral protein synthesis occurred. Xrn1 KO A549 cells were viable but nonpermissive for VACV; however, wild-type and mutant viruses replicated in triple-KO cells in which RNase L and PKR were also inactivated. Since KO of PKR and RNase L was sufficient to enable VACV replication in the absence of E3 or Xrn1, the poor replication of the decapping mutant, particularly in HAP1 DKO, cells indicated additional translational defects.
Importance: Viruses have evolved ways of preventing or counteracting the cascade of antiviral responses that double-stranded RNA (dsRNA) triggers in host cells. We showed that the dsRNA produced in excess in cells infected with a vaccinia virus (VACV) decapping enzyme mutant and by wild-type virus colocalized with the viral E3 protein in cytoplasmic viral factories. Novel human cell lines defective in either or both protein kinase R and RNase L dsRNA effector pathways and/or the cellular 5' exonuclease Xrn1 were prepared by CRISPR-Cas9 gene editing. Inactivation of both pathways was necessary and sufficient to allow full replication of the E3 mutant and reverse the defect cause by inactivation of Xrn1, whereas the decapping enzyme mutant still exhibited defects in gene expression. The study provided new insights into functions of the VACV proteins, and the well-characterized panel of CRISPR-Cas9-modified human cell lines should have broad applicability for studying innate dsRNA pathways.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures



Similar articles
-
Cellular 5'-3' mRNA exonuclease Xrn1 controls double-stranded RNA accumulation and anti-viral responses.Cell Host Microbe. 2015 Mar 11;17(3):332-344. doi: 10.1016/j.chom.2015.02.003. Cell Host Microbe. 2015. PMID: 25766294 Free PMC article.
-
Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice.J Virol. 2011 Jan;85(1):550-67. doi: 10.1128/JVI.00254-10. Epub 2010 Oct 13. J Virol. 2011. PMID: 20943971 Free PMC article.
-
Host species-specific activity of the poxvirus PKR inhibitors E3 and K3 mediate host range function.J Virol. 2024 Nov 19;98(11):e0133124. doi: 10.1128/jvi.01331-24. Epub 2024 Oct 31. J Virol. 2024. PMID: 39480085 Free PMC article.
-
Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses.Cell Host Microbe. 2015 Mar 11;17(3):320-331. doi: 10.1016/j.chom.2015.02.002. Cell Host Microbe. 2015. PMID: 25766293 Free PMC article.
-
The interferon system and vaccinia virus evasion mechanisms.J Interferon Cytokine Res. 2009 Sep;29(9):581-98. doi: 10.1089/jir.2009.0073. J Interferon Cytokine Res. 2009. PMID: 19708815 Review.
Cited by
-
Single Immunization with Recombinant ACAM2000 Vaccinia Viruses Expressing the Spike and the Nucleocapsid Proteins Protects Hamsters against SARS-CoV-2-Caused Clinical Disease.J Virol. 2022 May 11;96(9):e0038922. doi: 10.1128/jvi.00389-22. Epub 2022 Apr 12. J Virol. 2022. PMID: 35412347 Free PMC article.
-
Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus.Ann N Y Acad Sci. 2019 Feb;1438(1):18-29. doi: 10.1111/nyas.14000. Epub 2019 Jan 15. Ann N Y Acad Sci. 2019. PMID: 30644558 Free PMC article.
-
Activation of the antiviral factor RNase L triggers translation of non-coding mRNA sequences.Nucleic Acids Res. 2021 Jun 21;49(11):6007-6026. doi: 10.1093/nar/gkab036. Nucleic Acids Res. 2021. PMID: 33556964 Free PMC article.
-
Human Host Range Restriction of the Vaccinia Virus C7/K1 Double Deletion Mutant Is Mediated by an Atypical Mode of Translation Inhibition.J Virol. 2018 Nov 12;92(23):e01329-18. doi: 10.1128/JVI.01329-18. Print 2018 Dec 1. J Virol. 2018. PMID: 30209174 Free PMC article.
-
A Poxvirus Decapping Enzyme Colocalizes with Mitochondria To Regulate RNA Metabolism and Translation and Promote Viral Replication.mBio. 2022 Jun 28;13(3):e0030022. doi: 10.1128/mbio.00300-22. Epub 2022 Apr 18. mBio. 2022. PMID: 35435699 Free PMC article.
References
-
- DeWitte-Orr SJ, Mossman KL. 2010. dsRNA and the innate antiviral immune response. Future Virol 5:325–341. doi: 10.2217/fvl.10.18. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
