

A Novel Non-Replication-Competent Cytomegalovirus Capsid Mutant Vaccine Strategy Is Effective in Reducing Congenital Infection
- PMID: 27334585
- PMCID: PMC4988156
- DOI: 10.1128/JVI.00283-16
A Novel Non-Replication-Competent Cytomegalovirus Capsid Mutant Vaccine Strategy Is Effective in Reducing Congenital Infection
Abstract
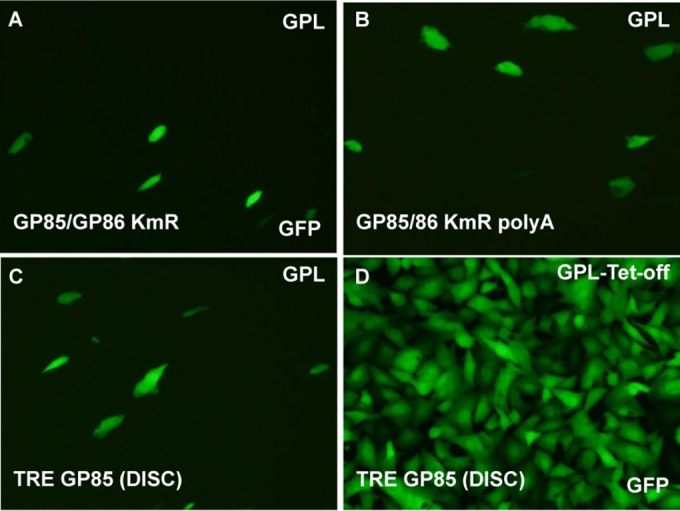
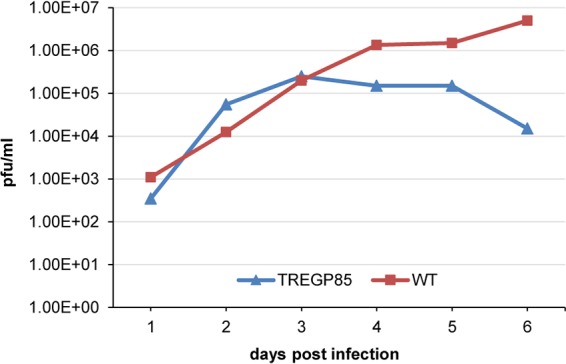
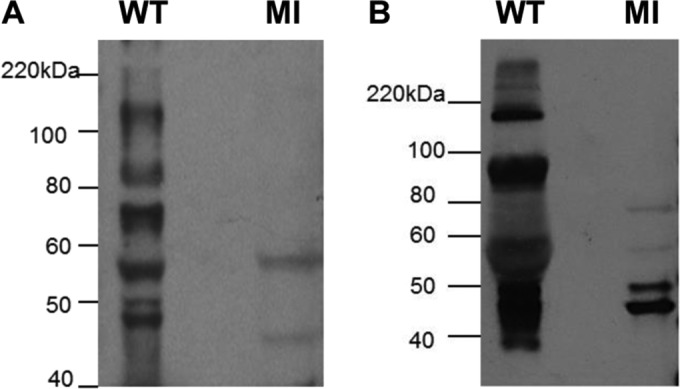
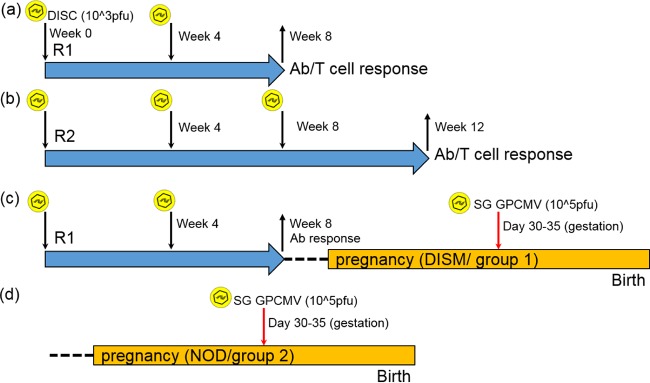
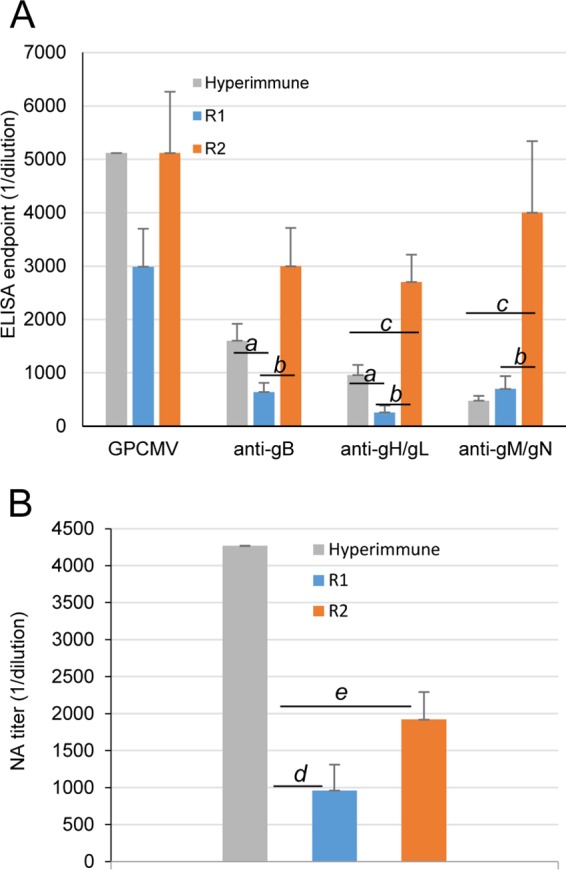
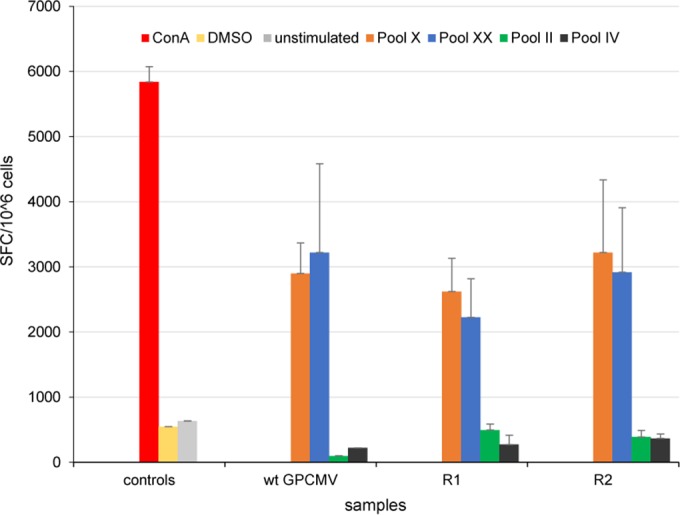
Congenital cytomegalovirus (CMV) infection is a leading cause of mental retardation and deafness in newborns. The guinea pig is the only small animal model for congenital CMV infection. A novel CMV vaccine was investigated as an intervention strategy against congenital guinea pig cytomegalovirus (GPCMV) infection. In this disabled infectious single-cycle (DISC) vaccine strategy, a GPCMV mutant virus was used that lacked the ability to express an essential capsid gene (the UL85 homolog GP85) except when grown on a complementing cell line. In vaccinated animals, the GP85 mutant virus (GP85 DISC) induced an antibody response to important glycoprotein complexes considered neutralizing target antigens (gB, gH/gL/gO, and gM/gN). The vaccine also generated a T cell response to the pp65 homolog (GP83), determined via a newly established guinea pig gamma interferon enzyme-linked immunosorbent spot assay. In a congenital infection protection study, GP85 DISC-vaccinated animals and a nonvaccinated control group were challenged during pregnancy with wild-type GPCMV (10(5) PFU). The pregnant animals carried the pups to term, and viral loads in target organs of pups were analyzed. Based on live pup births in the vaccinated and control groups (94.1% versus 63.6%), the vaccine was successful in reducing mortality (P = 0.0002). Additionally, pups from the vaccinated group had reduced CMV transmission, with 23.5% infected target organs versus 75.9% in the control group. Overall, these preliminary studies indicate that a DISC CMV vaccine strategy has the ability to induce an immune response similar to that of natural virus infection but has the increased safety of a non-replication-competent virus, which makes this approach attractive as a CMV vaccine strategy.
Importance: Congenital CMV infection is a leading cause of mental retardation and deafness in newborns. An effective vaccine against CMV remains an elusive goal despite over 50 years of CMV research. The guinea pig, with a placenta structure similar to that in humans, is the only small animal model for congenital CMV infection and recapitulates disease symptoms (e.g., deafness) in newborn pups. In this report, a novel vaccine strategy against congenital guinea pig cytomegalovirus (GPCMV) infection was developed, characterized, and tested for efficacy. This disabled infectious single-cycle (DISC) vaccine strategy induced a neutralizing antibody or a T cell response to important target antigens. In a congenital infection protection study, animals were protected against CMV in comparison to the nonvaccinated group (52% reduction of transmission). This novel vaccine was more effective than previously tested gB-based vaccines and most other strategies involving live virus vaccines. Overall, the DISC vaccine is a safe and promising approach against congenital CMV infection.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures

References
-
- Pass RF. 1996. Immunization strategy for prevention of congenital cytomegalovirus infection. Infect Agents Dis 5:240–244. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
