

The Ensembl gene annotation system
- PMID: 27337980
- PMCID: PMC4919035
- DOI: 10.1093/database/baw093
The Ensembl gene annotation system
Abstract
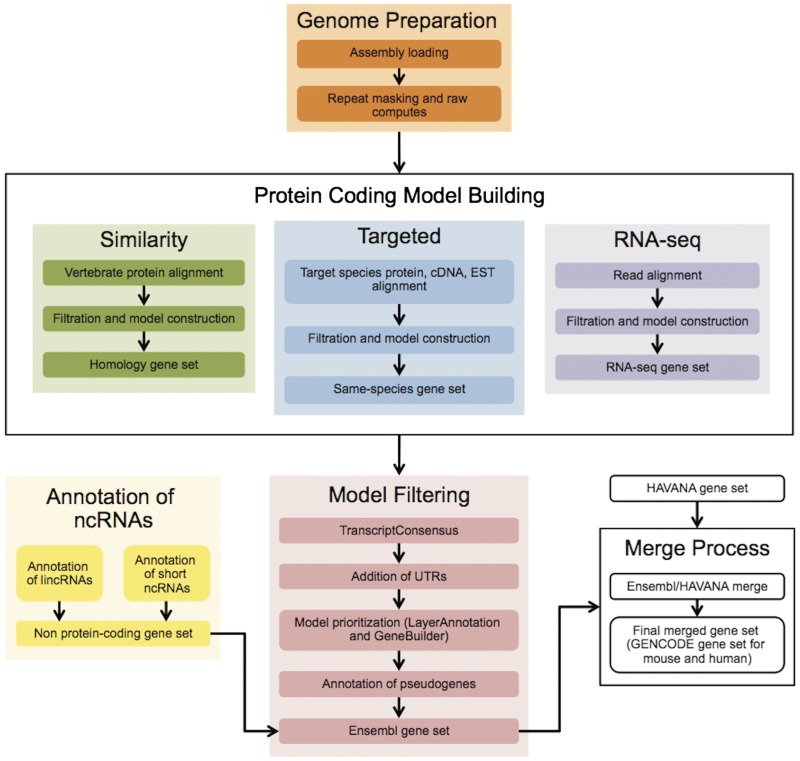
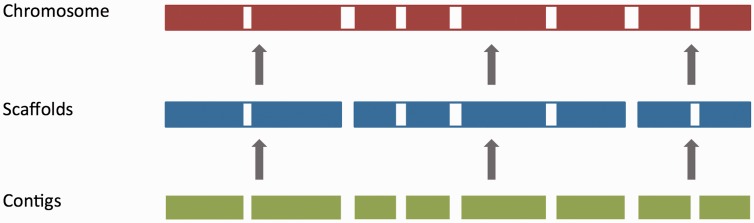
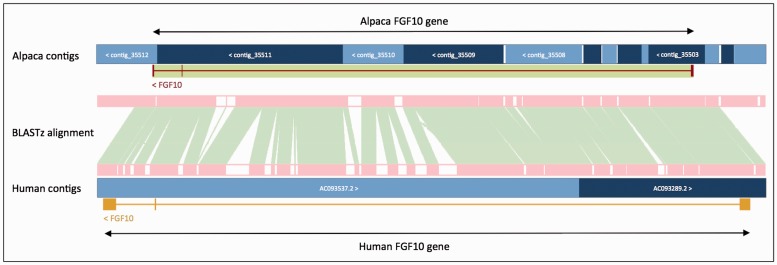
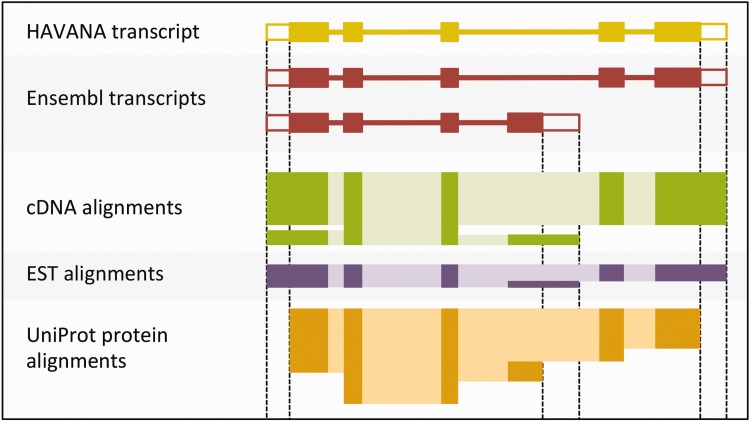
The Ensembl gene annotation system has been used to annotate over 70 different vertebrate species across a wide range of genome projects. Furthermore, it generates the automatic alignment-based annotation for the human and mouse GENCODE gene sets. The system is based on the alignment of biological sequences, including cDNAs, proteins and RNA-seq reads, to the target genome in order to construct candidate transcript models. Careful assessment and filtering of these candidate transcripts ultimately leads to the final gene set, which is made available on the Ensembl website. Here, we describe the annotation process in detail.Database URL: http://www.ensembl.org/index.html.
© The Author(s) 2016. Published by Oxford University Press.
Figures
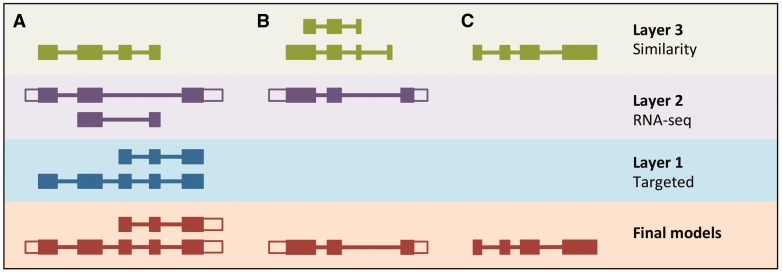
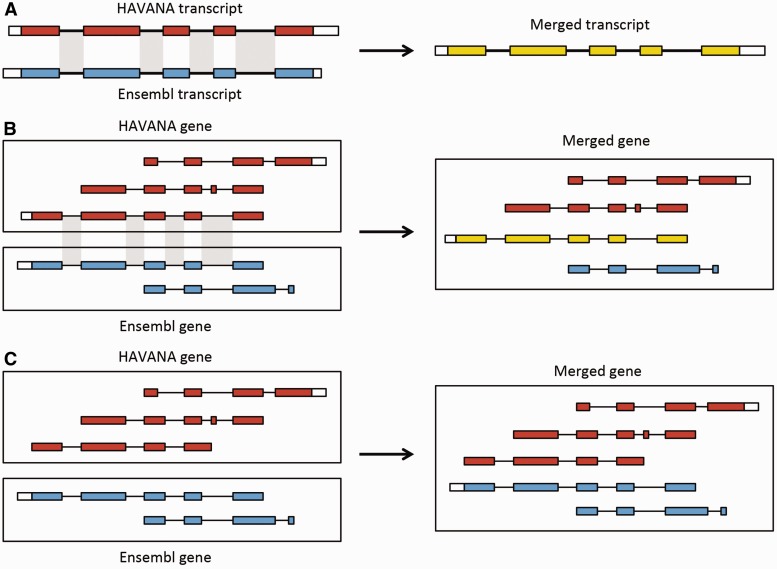
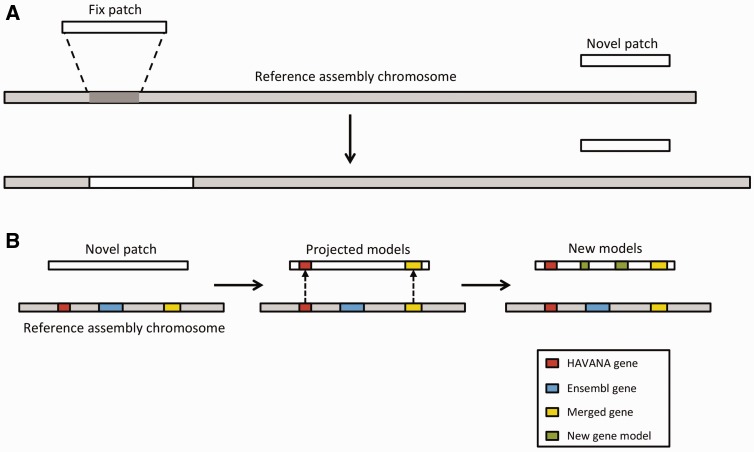
References
-
- Brent M.R. (2005) Genome annotation past, present, and future: how to define an ORF at each locus. Genome Res., 15, 1777–1786. - PubMed
MeSH terms
Grants and funding
- BBS/B/13446/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/E011640/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U54 HG004555/HG/NHGRI NIH HHS/United States
- BB/I025360/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M011615/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT095908/WT_/Wellcome Trust/United Kingdom
- BB/I025506/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M018458/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- WT098051/WT_/Wellcome Trust/United Kingdom
- U41 HG007234/HG/NHGRI NIH HHS/United States
- R01 HD074078/HD/NICHD NIH HHS/United States
- BB/I025360/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/B/13470/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M011461/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/K009524/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
