

Dihydromunduletone Is a Small-Molecule Selective Adhesion G Protein-Coupled Receptor Antagonist
- PMID: 27338081
- PMCID: PMC4998661
- DOI: 10.1124/mol.116.104828
Dihydromunduletone Is a Small-Molecule Selective Adhesion G Protein-Coupled Receptor Antagonist
Abstract
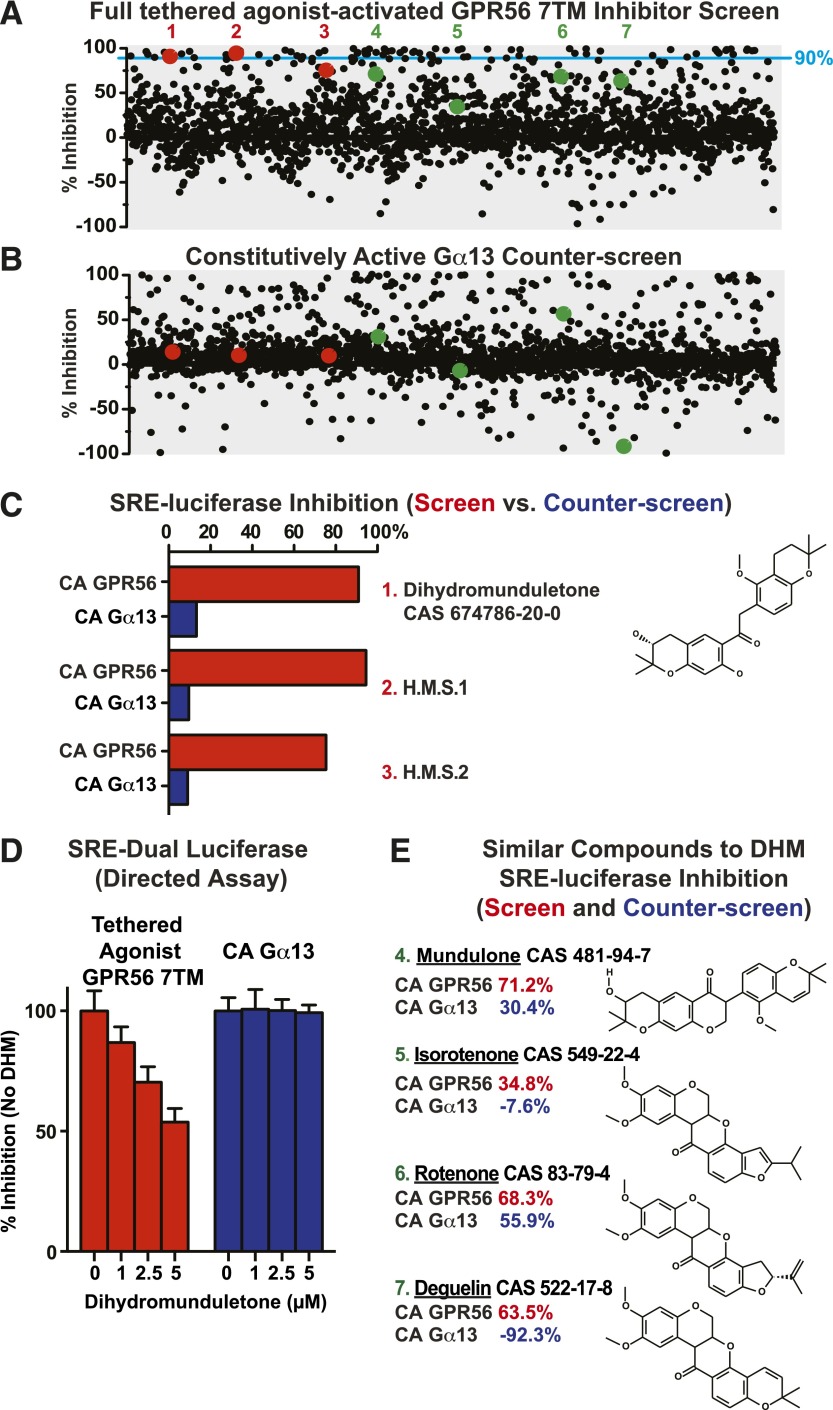
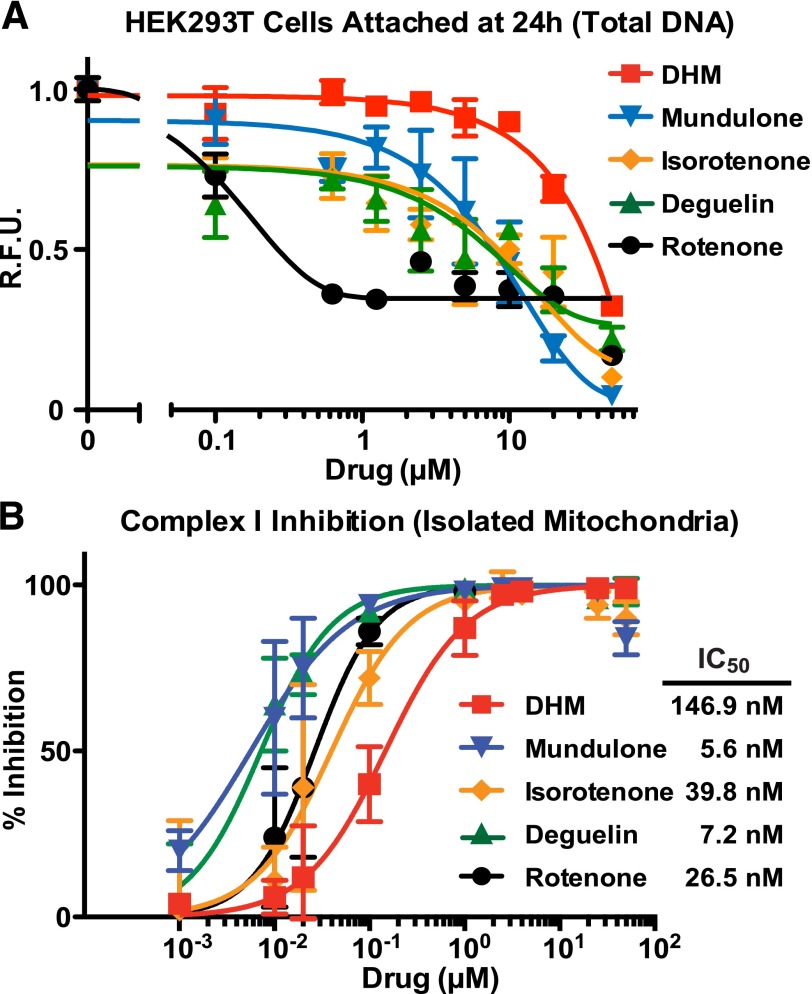
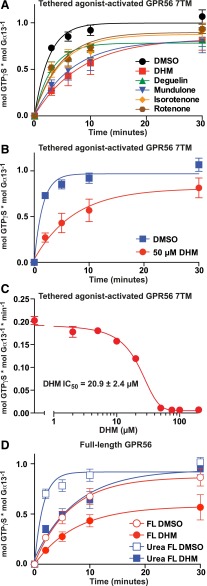
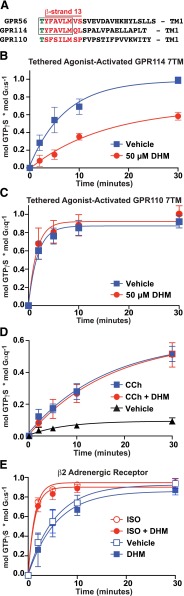
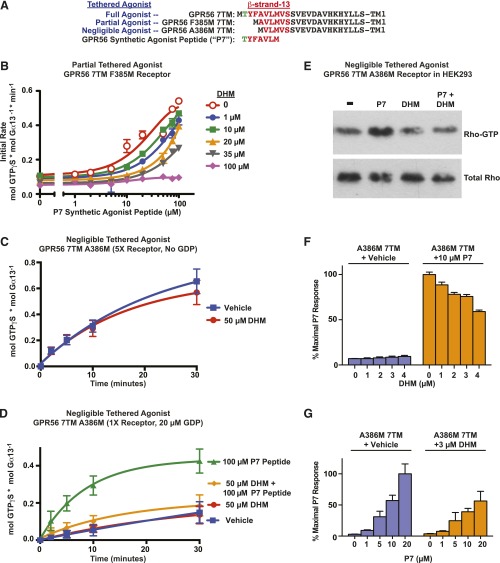
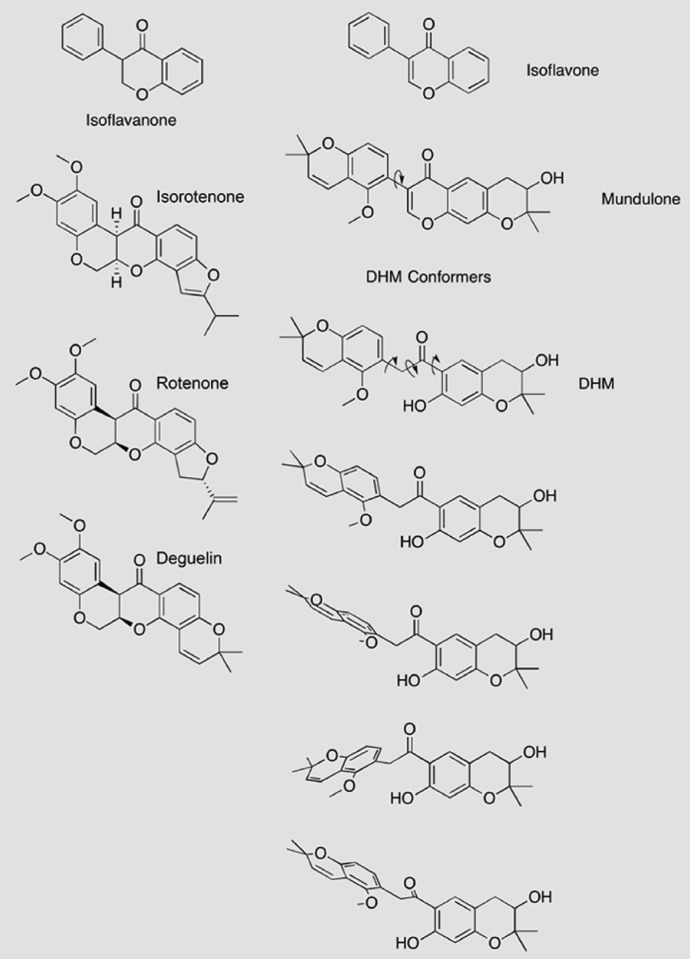
Adhesion G protein-coupled receptors (aGPCRs) have emerging roles in development and tissue maintenance and is the most prevalent GPCR subclass mutated in human cancers, but to date, no drugs have been developed to target them in any disease. aGPCR extracellular domains contain a conserved subdomain that mediates self-cleavage proximal to the start of the 7-transmembrane domain (7TM). The two receptor protomers, extracellular domain and amino terminal fragment (NTF), and the 7TM or C-terminal fragment remain noncovalently bound at the plasma membrane in a low-activity state. We recently demonstrated that NTF dissociation liberates the 7TM N-terminal stalk, which acts as a tethered-peptide agonist permitting receptor-dependent heterotrimeric G protein activation. In many cases, natural aGPCR ligands are extracellular matrix proteins that dissociate the NTF to reveal the tethered agonist. Given the perceived difficulty in modifying extracellular matrix proteins to create aGPCR probes, we developed a serum response element (SRE)-luciferase-based screening approach to identify GPR56/ADGRG1 small-molecule inhibitors. A 2000-compound library comprising known drugs and natural products was screened for GPR56-dependent SRE activation inhibitors that did not inhibit constitutively active Gα13-dependent SRE activation. Dihydromunduletone (DHM), a rotenoid derivative, was validated using cell-free aGPCR/heterotrimeric G protein guanosine 5'-3-O-(thio)triphosphate binding reconstitution assays. DHM inhibited GPR56 and GPR114/ADGRG5, which have similar tethered agonists, but not the aGPCR GPR110/ADGRF1, M3 muscarinic acetylcholine, or β2 adrenergic GPCRs. DHM inhibited tethered peptide agonist-stimulated and synthetic peptide agonist-stimulated GPR56 but did not inhibit basal activity, demonstrating that it antagonizes the peptide agonist. DHM is a novel aGPCR antagonist and potentially useful chemical probe that may be developed as a future aGPCR therapeutic.
Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
References
-
- Bindslev N(2008) The Schild against other theories, in Drug-Acceptor Interactions pp 283–298, Coaction Publishing, Stockholm.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
