

Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies
- PMID: 27339098
- PMCID: PMC5707186
- DOI: 10.1016/j.jmoldx.2016.02.003
Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies
Abstract
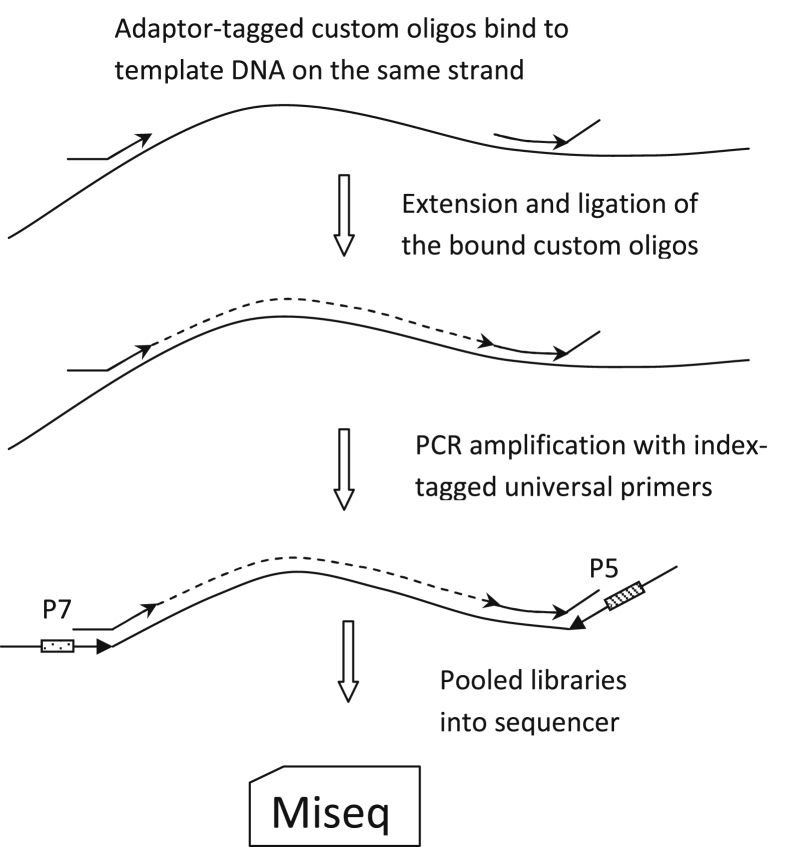
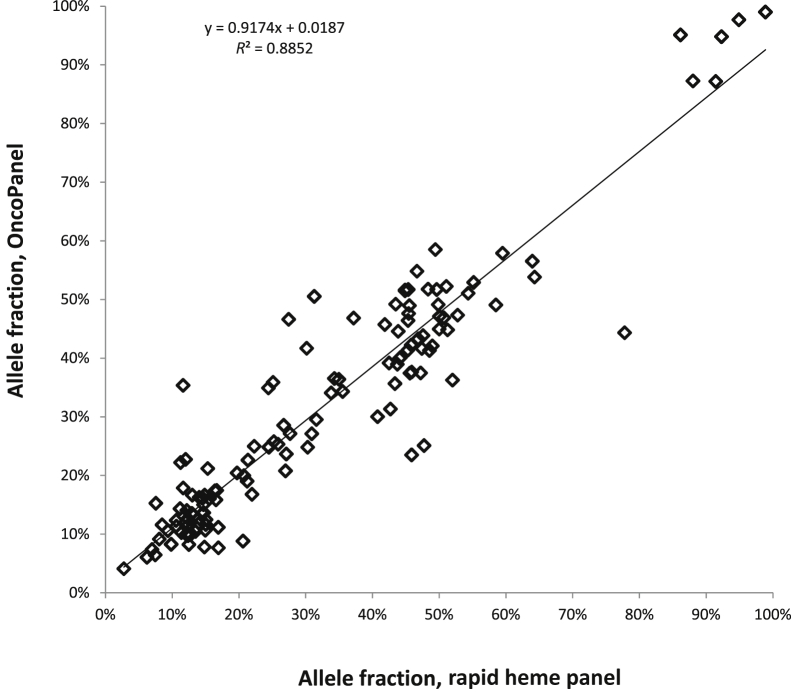
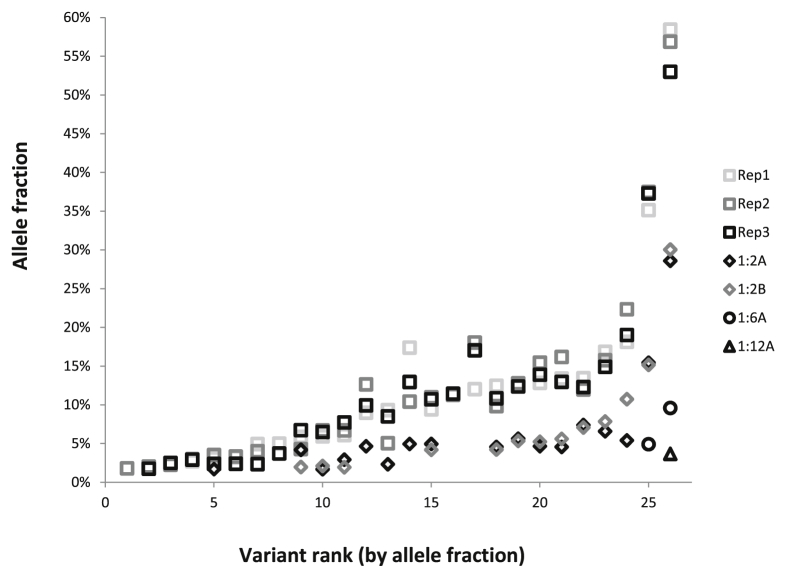
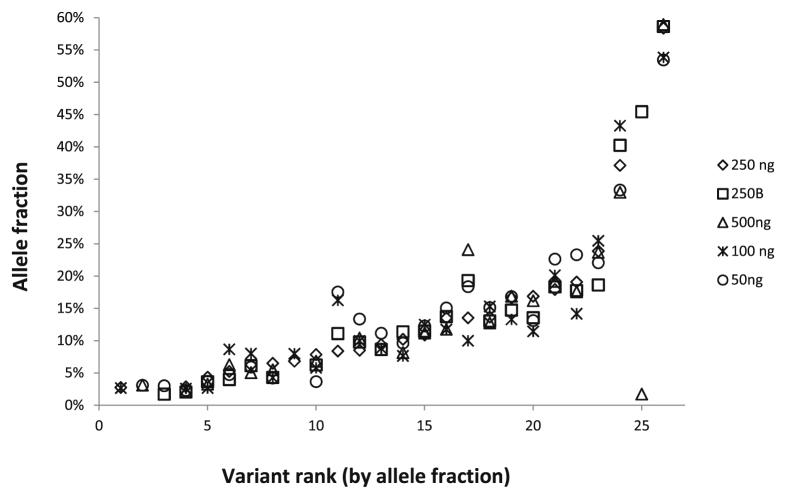
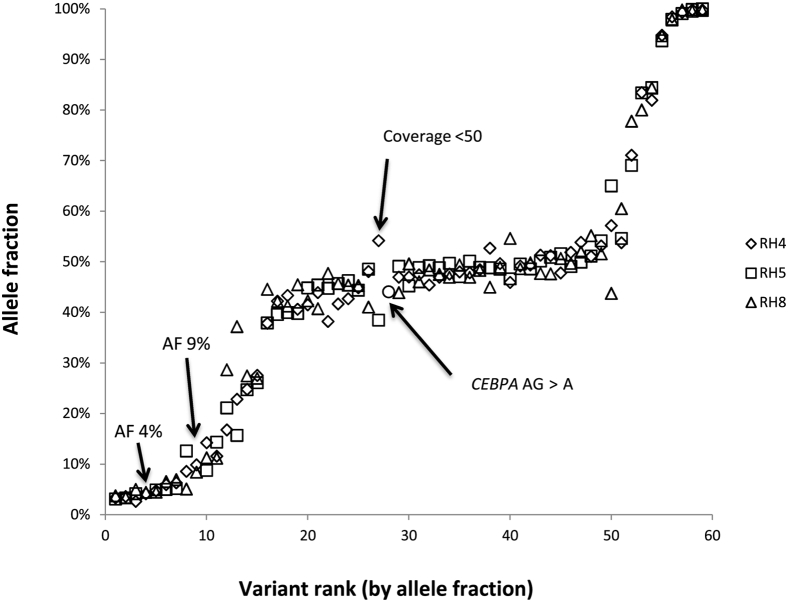
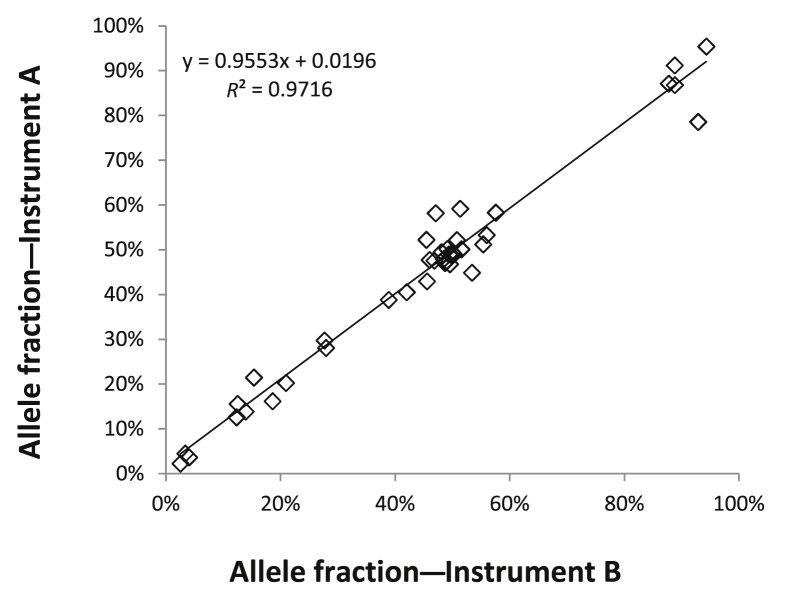
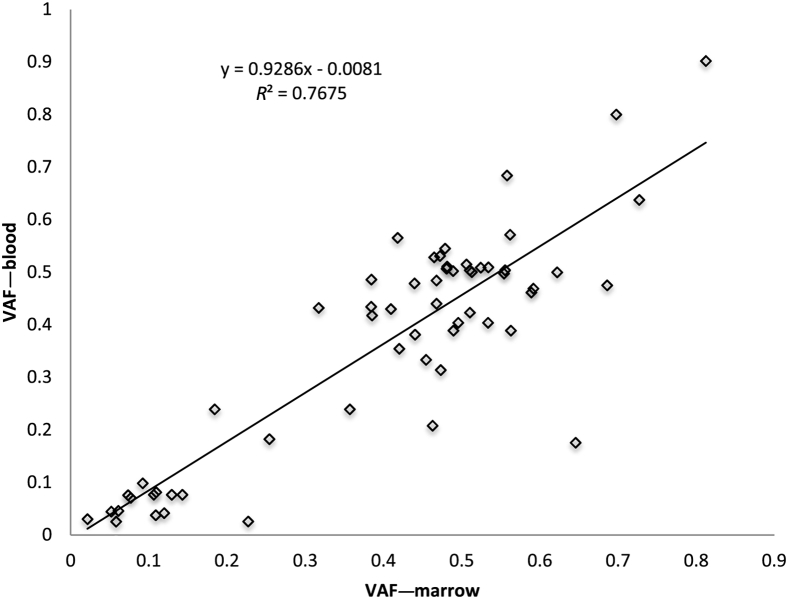
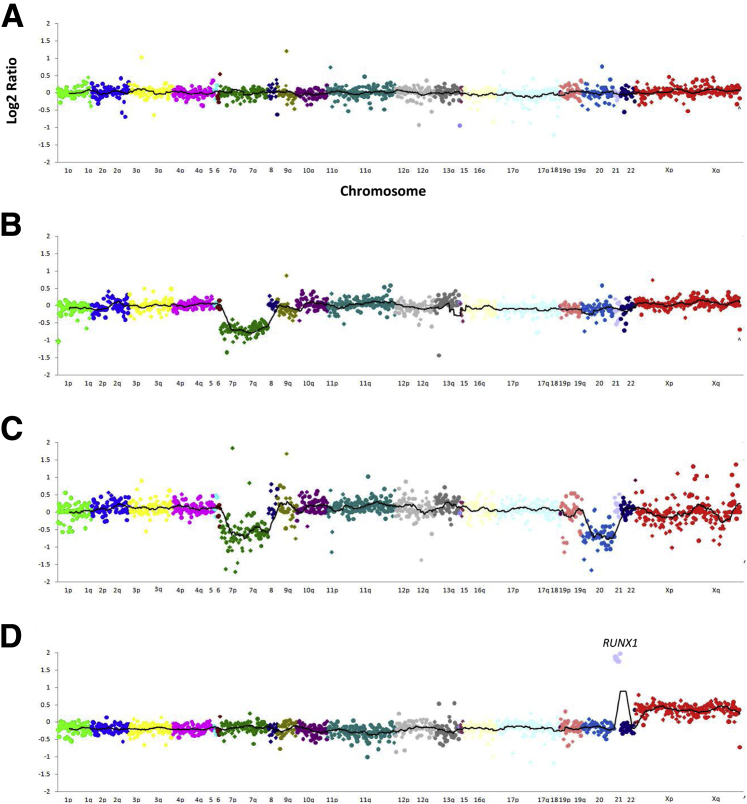
Targeted next-generation sequencing panels to identify genetic alterations in cancers are increasingly becoming an integral part of clinical practice. We report here the design, validation, and implementation of a comprehensive 95-gene next-generation sequencing panel targeted for hematologic malignancies that we named rapid heme panel. Rapid heme panel is amplicon based and covers hotspot regions of oncogenes and most of the coding regions of tumor suppressor genes. It is composed of 1330 amplicons and covers 175 kb of genomic sequence in total. Rapid heme panel's average coverage is 1500× with <5% of the amplicons with <50× coverage, and it reproducibly detects single nucleotide variants and small insertions/deletions at allele frequencies of ≥5%. Comparison with a capture-based next-generation sequencing assay showed that there is >95% concordance among a wide array of variants across a range of allele frequencies. Read count analyses that used rapid heme panel showed high concordance with karyotypic results when tumor content was >30%. The average turnaround time was 7 days over a 6-month span with an average volume of ≥40 specimens per week and a low sample fail rate (<1%), demonstrating its suitability for clinical application.
Copyright © 2016 American Society for Investigative Pathology and the Association for Molecular Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Metzker M.L. Sequencing technologies — the next generation. Nat Rev Genet. 2010;11:31–46. - PubMed
-
- Lindsley R.C., Ebert B.L. The biology and clinical impact of genetic lesions in myeloid malignancies. Blood. 2013;122:3741–3748. - PubMed
-
- Lundberg P., Karow A., Nienhold R., Looser R., Hao-Shen H., Nissen I., Girsberger S., Lehmann T., Passweg J., Stern M., Beisel C., Kralovics R., Skoda R.C. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123:2220–2228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
