

A survey of the sorghum transcriptome using single-molecule long reads
- PMID: 27339290
- PMCID: PMC4931028
- DOI: 10.1038/ncomms11706
A survey of the sorghum transcriptome using single-molecule long reads
Abstract
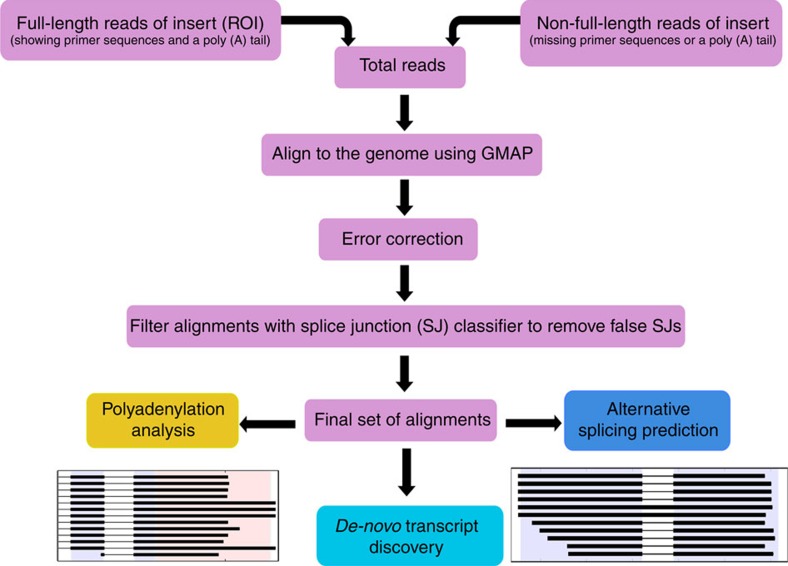
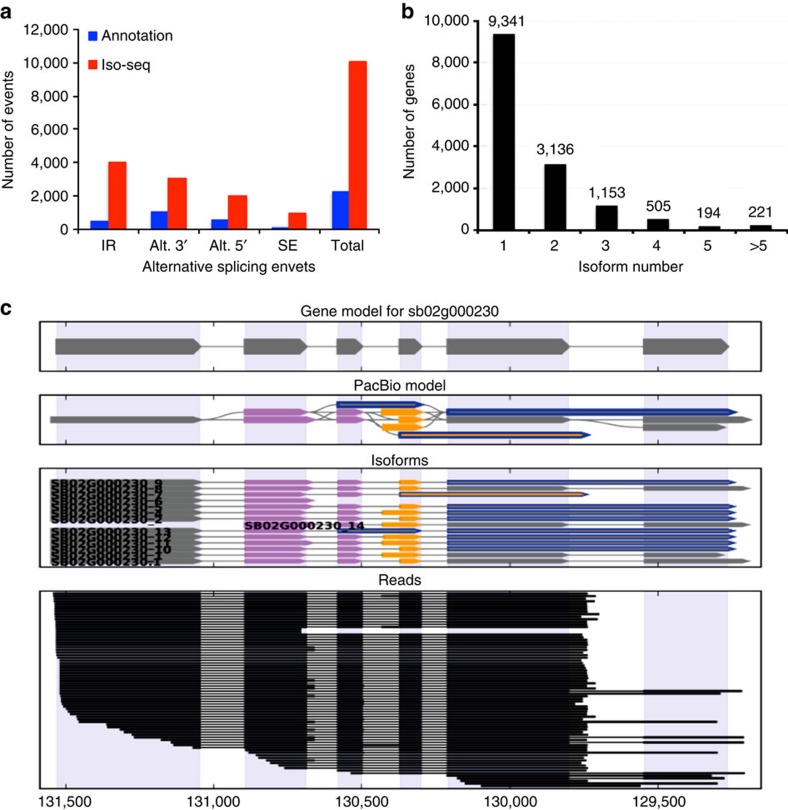
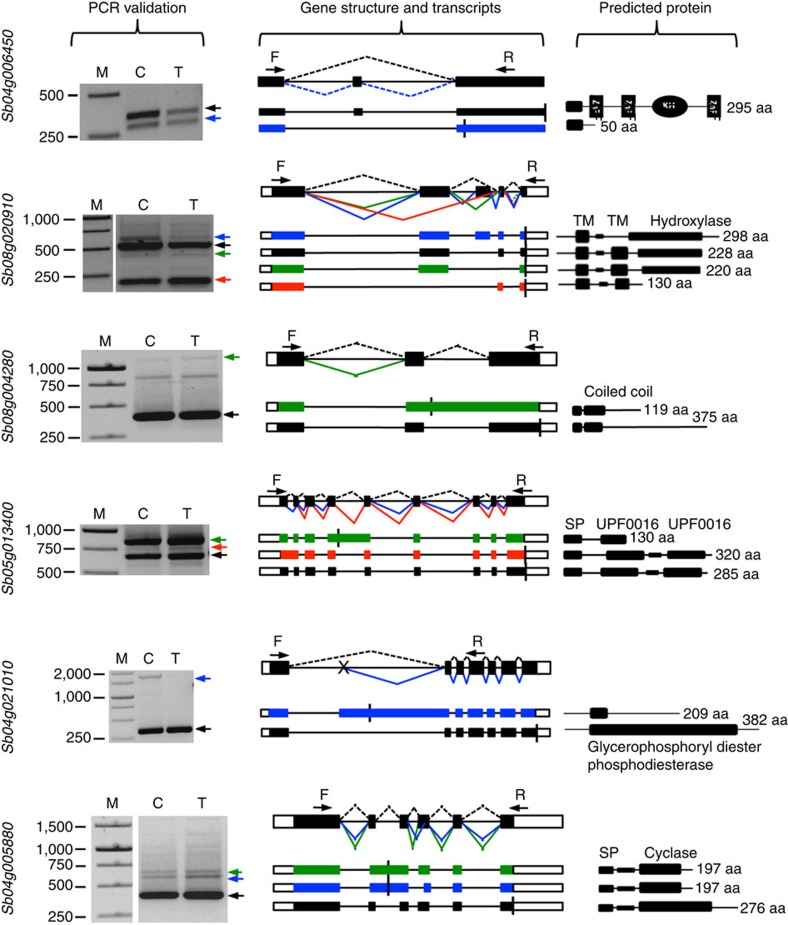
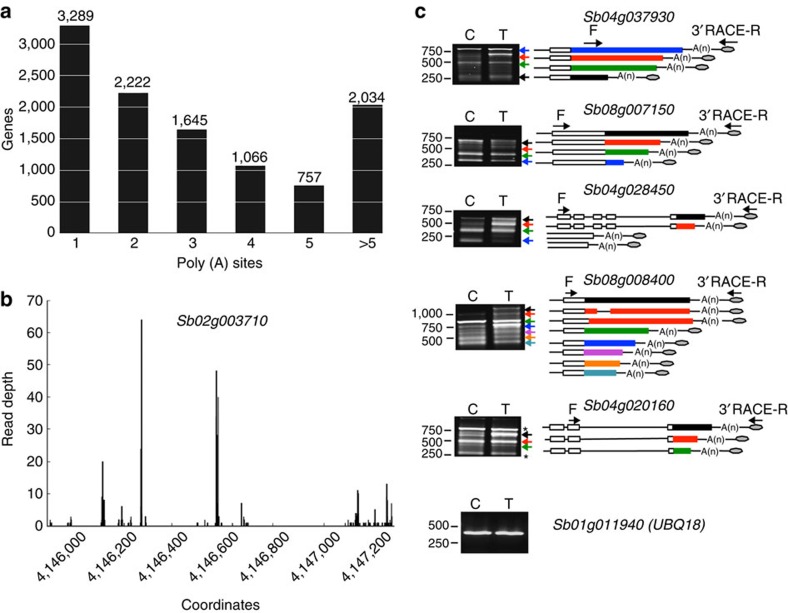
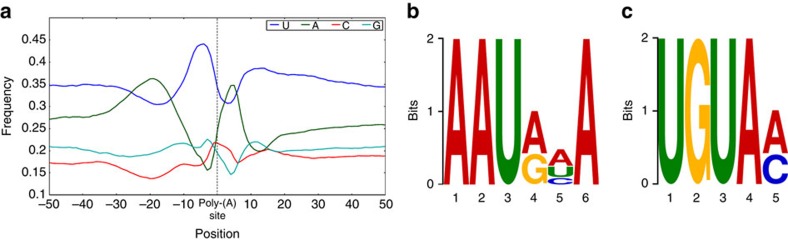
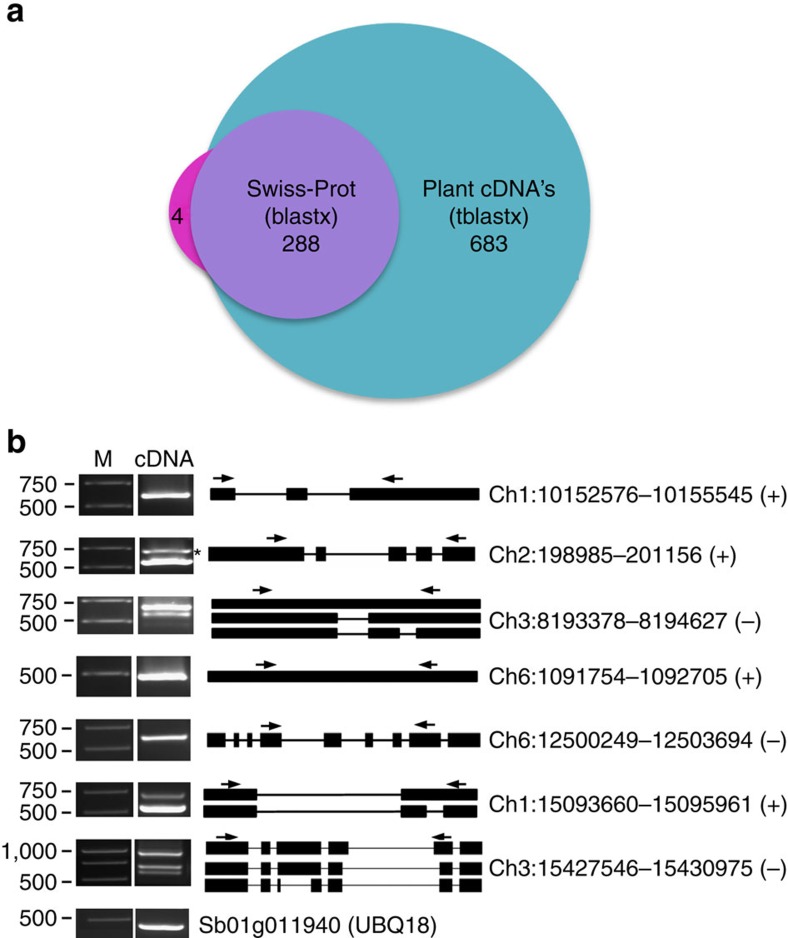
Alternative splicing and alternative polyadenylation (APA) of pre-mRNAs greatly contribute to transcriptome diversity, coding capacity of a genome and gene regulatory mechanisms in eukaryotes. Second-generation sequencing technologies have been extensively used to analyse transcriptomes. However, a major limitation of short-read data is that it is difficult to accurately predict full-length splice isoforms. Here we sequenced the sorghum transcriptome using Pacific Biosciences single-molecule real-time long-read isoform sequencing and developed a pipeline called TAPIS (Transcriptome Analysis Pipeline for Isoform Sequencing) to identify full-length splice isoforms and APA sites. Our analysis reveals transcriptome-wide full-length isoforms at an unprecedented scale with over 11,000 novel splice isoforms. Additionally, we uncover APA of ∼11,000 expressed genes and more than 2,100 novel genes. These results greatly enhance sorghum gene annotations and aid in studying gene regulation in this important bioenergy crop. The TAPIS pipeline will serve as a useful tool to analyse Iso-Seq data from any organism.
Figures
References
-
- Elkon R., Ugalde A. P. & Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. Genet. 14, 496–506 (2013). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
