

Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences
- PMID: 27340661
- PMCID: PMC4908263
- DOI: 10.1155/2016/5258727
Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences
Abstract
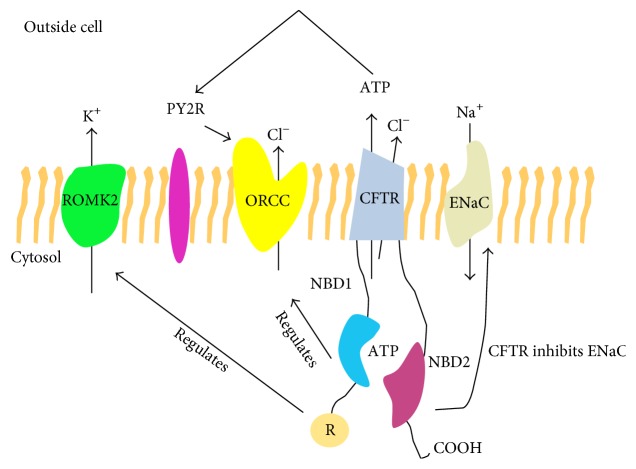
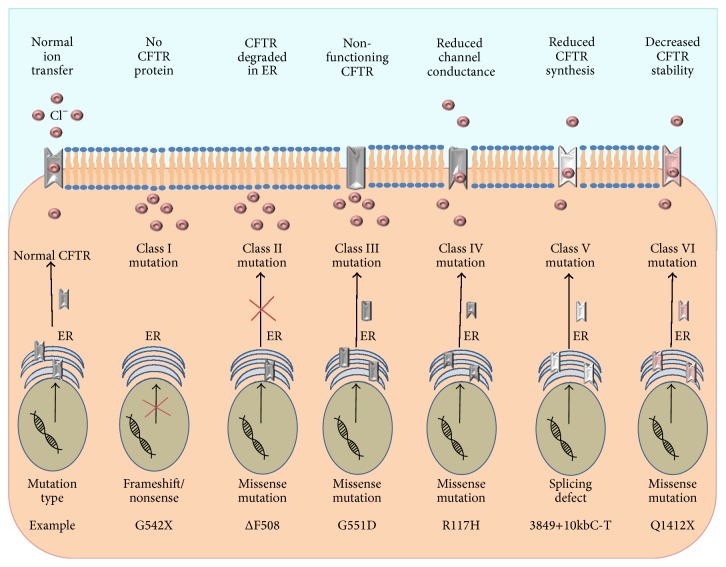
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The resultant characteristic ion transport defect results in decreased mucociliary clearance, bacterial colonisation, and chronic neutrophil-dominated inflammation. Much knowledge surrounding the pathophysiology of the disease has been gained through the generation of animal models, despite inherent limitations in each. The failure of certain mouse models to recapitulate the phenotypic manifestations of human disease has initiated the generation of larger animals in which to study CF, including the pig and the ferret. This review will summarise the basic phenotypes of three animal models and describe the contributions of such animal studies to our current understanding of CF.
Figures
References
-
- The Cystic Fibrosis Registry of Ireland. 2012 Annual Report. Dublin, Ireland: The Cystic Fibrosis Registry of Ireland; 2012.
-
- Cystic Fibrosis Foundation. Patient Registry—Annual Data Report 2012. Bethesda, Md, USA: Cystic Fibrosis Foundation; 2012.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
