

A mechanism for 1,4-Benzoquinone-induced genotoxicity
- PMID: 27340773
- PMCID: PMC5216808
- DOI: 10.18632/oncotarget.10184
A mechanism for 1,4-Benzoquinone-induced genotoxicity
Abstract
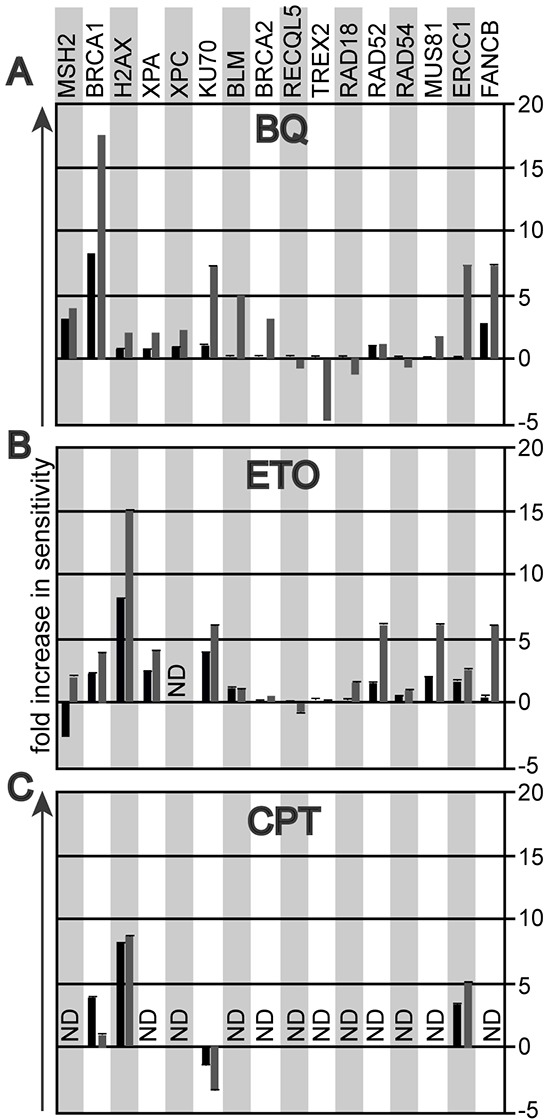
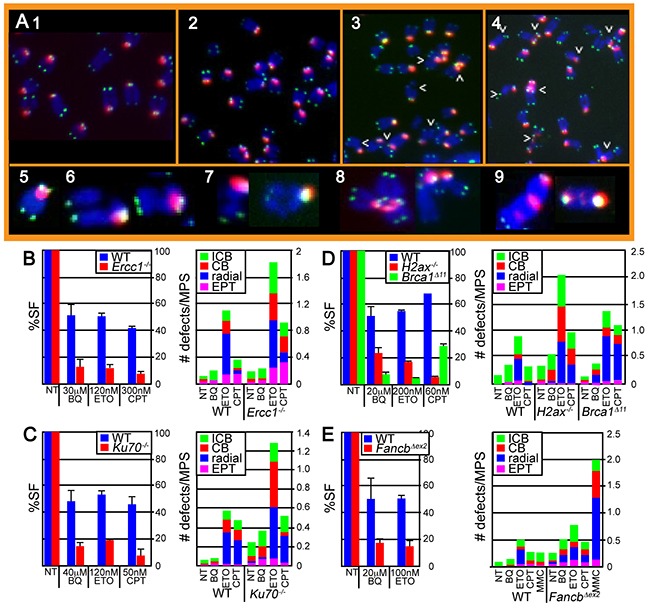
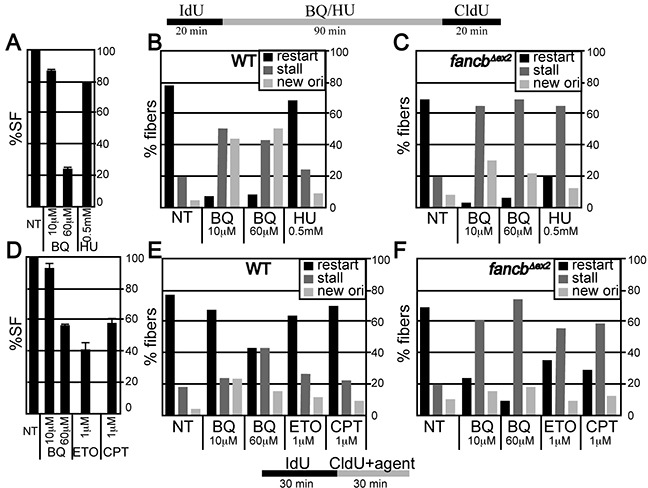
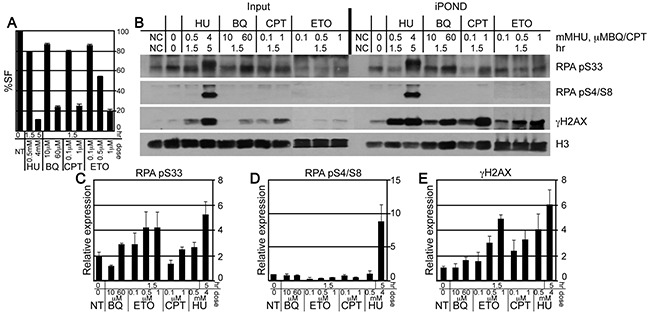
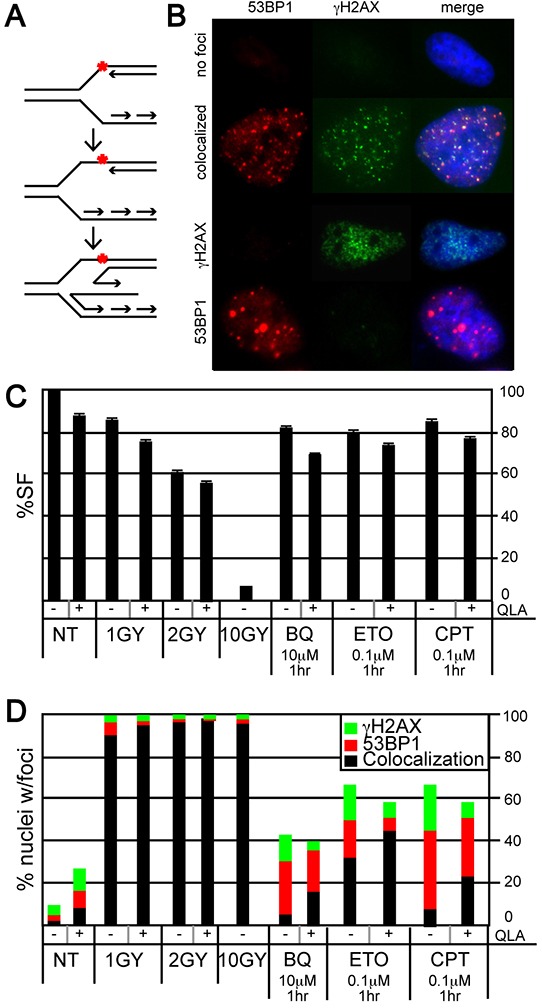
Benzene is a common environmental toxin and its metabolite, 1-4-Benzoquinone (BQ) causes hematopoietic cancers like myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML). BQ has not been comprehensively assessed for its impact on genome maintenance, limiting our understanding of the true health risks associated with benzene exposure and our ability to identify people with increased sensitivity to this genotoxin. Here we analyze the impact BQ exposure has on wild type and DNA repair-defective mouse embryonic stem (ES) cells and wild type human cells. We find that double strand break (DSB) repair and replication fork maintenance pathways including homologous recombination (HR) and Fanconi anemia (FA) suppress BQ toxicity. BQ-induced damage efficiently stalls replication forks, yet poorly induces ATR/DNA-PKCS responses. Furthermore, the pattern of BQ-induced γH2AX and 53BP1foci is consistent with the formation of poly(ADP-ribose) polymerase 1 (PARP1)-stabilized regressed replication forks. At a biochemical level, BQ inhibited topoisomerase 1 (topo1)-mediated DNA ligation and nicking in vitro; thus providing mechanism for the cellular phenotype. These data are consistent with a model that proposes BQ interferes with type I topoisomerase's ability to maintain replication fork restart and progression leading to chromosomal instability that has the potential to cause hematopoietic cancers like MDS and AML.
Keywords: Fanconi anemia; double strand break repair; replication fork maintenance; type 1 topoisomerase.
Conflict of interest statement
The authors have no conflict of interest.
Figures

Similar articles
-
Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks.Mutat Res. 2014 Aug-Sep;766-767:66-72. doi: 10.1016/j.mrfmmm.2014.06.003. Epub 2014 Jun 22. Mutat Res. 2014. PMID: 25847274
-
Deletion of BRCA2 exon 27 causes defects in response to both stalled and collapsed replication forks.Mutat Res. 2014 Aug-Sep;766-767:66-72. doi: 10.1016/j.mrfmmm.2014.06.003. Epub 2014 Jun 22. Mutat Res. 2014. PMID: 25773776 Free PMC article.
-
Generation of phosphorylated histone H2AX by benzene metabolites.Toxicol In Vitro. 2008 Dec;22(8):1861-8. doi: 10.1016/j.tiv.2008.09.005. Epub 2008 Sep 18. Toxicol In Vitro. 2008. PMID: 18835433
-
BRCA2 functions: from DNA repair to replication fork stabilization.Endocr Relat Cancer. 2016 Oct;23(10):T1-T17. doi: 10.1530/ERC-16-0297. Epub 2016 Aug 16. Endocr Relat Cancer. 2016. PMID: 27530658 Review.
-
Homologous Recombination-Mediated DNA Repair and Implications for Clinical Treatment of Repair Defective Cancers.Methods Mol Biol. 2019;1999:3-29. doi: 10.1007/978-1-4939-9500-4_1. Methods Mol Biol. 2019. PMID: 31127567 Review.
Cited by
-
Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes.Cancers (Basel). 2021 Mar 24;13(7):1494. doi: 10.3390/cancers13071494. Cancers (Basel). 2021. PMID: 33804958 Free PMC article.
-
PTP4A3, A Novel Target Gene of HIF-1alpha, Participates in Benzene-Induced Cell Proliferation Inhibition and Apoptosis through PI3K/AKT Pathway.Int J Environ Res Public Health. 2020 Feb 1;17(3):910. doi: 10.3390/ijerph17030910. Int J Environ Res Public Health. 2020. PMID: 32024182 Free PMC article.
-
Establishment of a zebrafish hematological disease model induced by 1,4-benzoquinone.Dis Model Mech. 2019 Mar 28;12(3):dmm037903. doi: 10.1242/dmm.037903. Dis Model Mech. 2019. PMID: 30898970 Free PMC article.
-
Topoisomerase II contributes to DNA secondary structure-mediated double-stranded breaks.Nucleic Acids Res. 2020 Jul 9;48(12):6654-6671. doi: 10.1093/nar/gkaa483. Nucleic Acids Res. 2020. PMID: 32501506 Free PMC article.
-
Linking Benzene, in Utero Carcinogenicity and Fetal Hematopoietic Stem Cell Niches: A Mechanistic Review.Int J Mol Sci. 2023 Mar 28;24(7):6335. doi: 10.3390/ijms24076335. Int J Mol Sci. 2023. PMID: 37047305 Free PMC article. Review.
References
-
- Hayes RB, Songnian Y, Dosemeci M, Linet M. Benzene and lymphohematopoietic malignancies in humans. Am J Ind Med. 2001;40:117–126. - PubMed
-
- Rinsky RA, Smith AB, Hornung R, Filloon TG, Young RJ, Okun AH, Landrigan PJ. Benzene and leukemia An epidemiologic risk assessment. N Engl J Med. 1987;316:1044–1050. - PubMed
-
- Hartwig A. The role of DNA repair in benzene-induced carcinogenesis. Chem Biol Interact. 2010;184:269–272. - PubMed
-
- Aul C, Bowen DT, Yoshida Y. Pathogenesis etiology and epidemiology of myelodysplastic syndromes. Haematologica. 1998;83:71–86. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
