

Evidence of extensive non-allelic gene conversion among LTR elements in the human genome
- PMID: 27346230
- PMCID: PMC4921805
- DOI: 10.1038/srep28710
Evidence of extensive non-allelic gene conversion among LTR elements in the human genome
Abstract
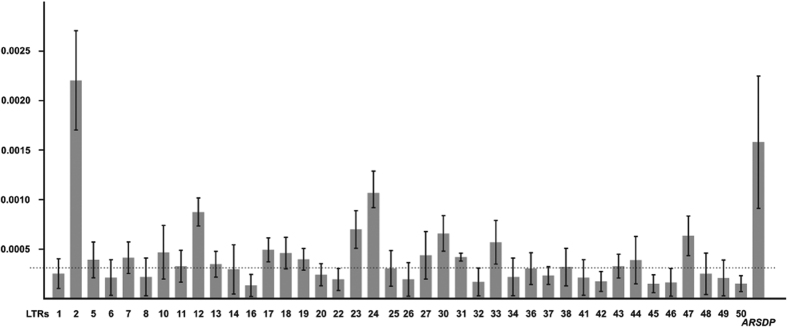
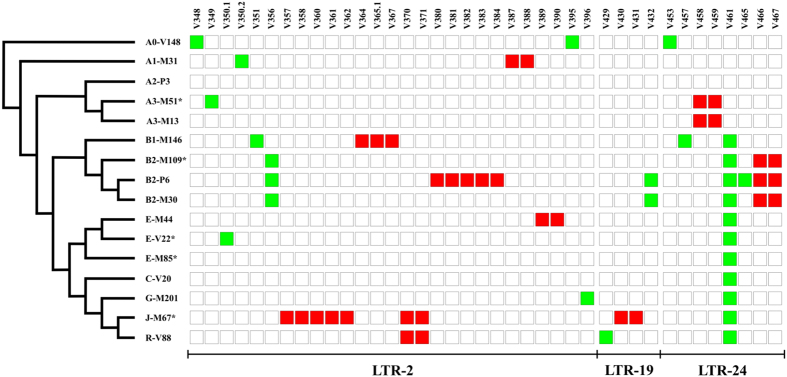
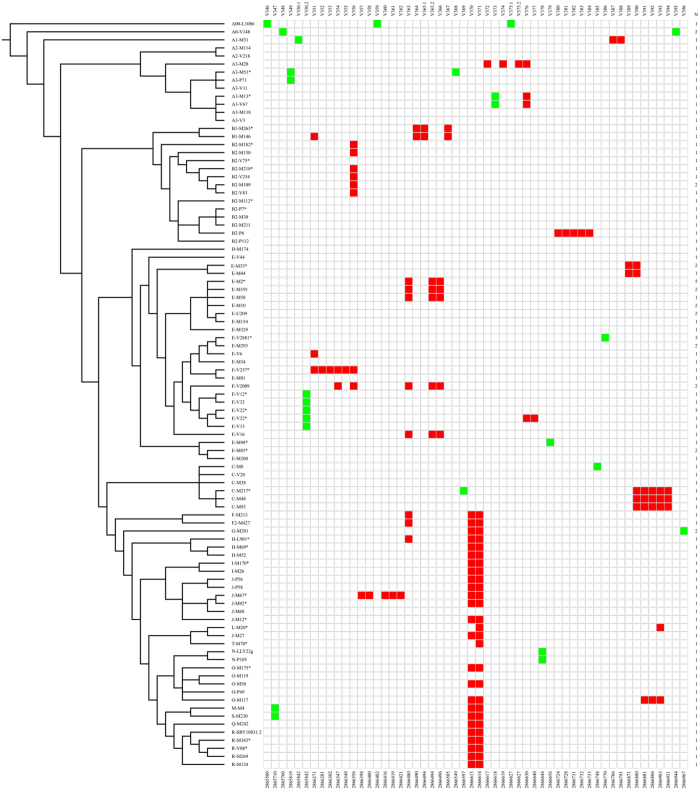
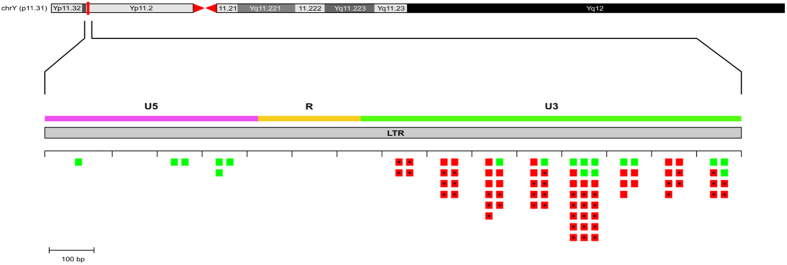
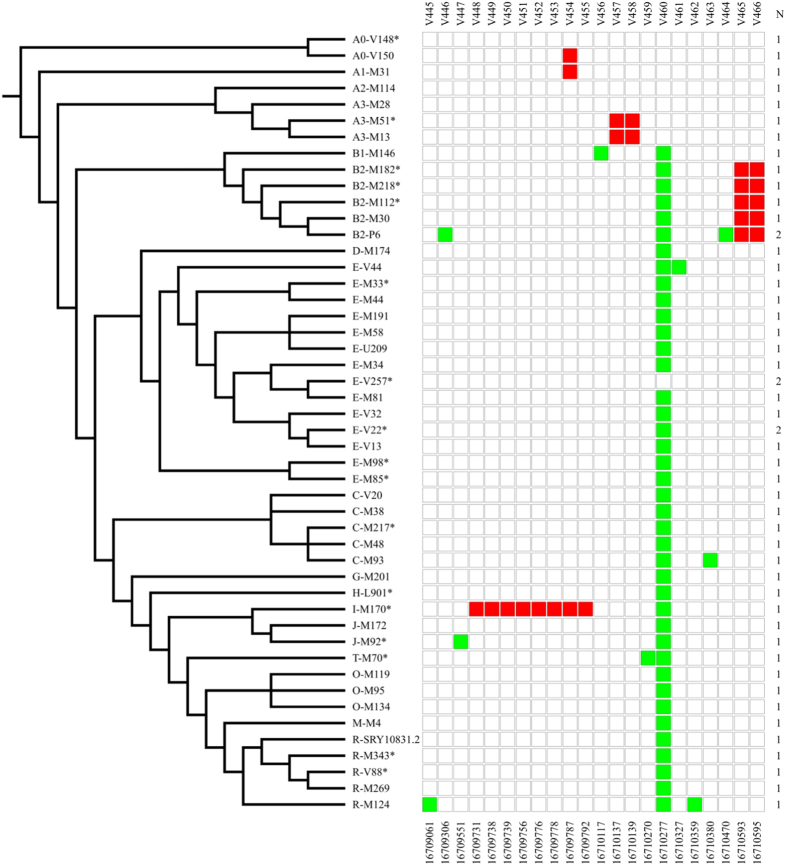
Long Terminal Repeats (LTRs) are nearly identical DNA sequences found at either end of Human Endogenous Retroviruses (HERVs). The high sequence similarity that exists among different LTRs suggests they could be substrate of ectopic gene conversion events. To understand the extent to which gene conversion occurs and to gain new insights into the evolutionary history of these elements in humans, we performed an intra-species phylogenetic study of 52 LTRs on different unrelated Y chromosomes. From this analysis, we obtained direct evidence that demonstrates the occurrence of ectopic gene conversion in several LTRs, with donor sequences located on both sex chromosomes and autosomes. We also found that some of these elements are characterized by an extremely high density of polymorphisms, showing one of the highest nucleotide diversities in the human genome, as well as a complex patchwork of sequences derived from different LTRs. Finally, we highlighted the limits of current short-read NGS studies in the analysis of genetic diversity of the LTRs in the human genome. In conclusion, our comparative re-sequencing analysis revealed that ectopic gene conversion is a common event in the evolution of LTR elements, suggesting complex genetic links among LTRs from different chromosomes.
Figures
References
-
- Kurth R. & Bannert N. Beneficial and detrimental effects of human endogenous retroviruses. Int. J. Cancer. 126, 306–314 (2010). - PubMed
-
- Feschotte C. & Gilbert C. Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13, 283–296 (2012). - PubMed
-
- Stoye J. P. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat. Rev. Microbiol. 10, 395–406 (2012). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
