

BRAFV600E-dependent Mcl-1 stabilization leads to everolimus resistance in colon cancer cells
- PMID: 27351224
- PMCID: PMC5216972
- DOI: 10.18632/oncotarget.10277
BRAFV600E-dependent Mcl-1 stabilization leads to everolimus resistance in colon cancer cells
Abstract
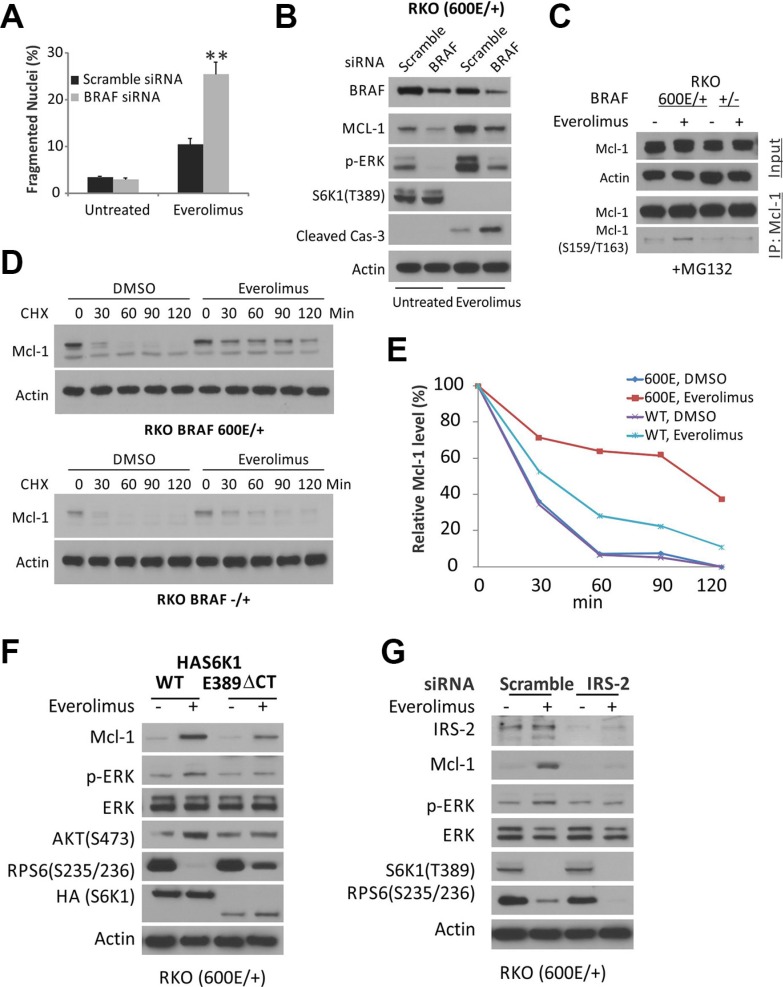
mTOR activation is commonly caused by oncogenic mutations in RAS/RAF/MAPK and PI3K/AKT pathways, and promotes cancer progression and therapeutic resistance. However, mTOR inhibitors show limited single agent efficacy in patients. mTOR inhibitors suppress tumor cell growth and angiogenesis, and have recently been shown to induce death receptor/FADD-dependent apoptosis in colon cancers. Using a panel of BRAF V600E and WT colorectal cancer cell lines and in vitro selected resistant culture, and xenograft models, we demonstrate here that BRAFV600E confers resistance to mTOR inhibitors. Everolimus treatment disrupts the S6K1-IRS-2/PI3K negative feedback loop, leading to BRAF V600E-dependent activation of ERK and Mcl-1 stabilization in colon cancer cells, which in turn blocks the crosstalk from the death receptor to mitochondria. Co-treatment with inhibitors to Mcl-1, PI3K, RAF or MEK restores mTOR inhibitor-induced apoptosis by antagonizing Mcl-1 or abrogating ERK activation in BRAFV600E cells. Our findings provide a rationale for genotype-guided patient stratification and potential drug combinations to prevent or mitigate undesired activation of survival pathways induced by mTOR inhibitors.
Keywords: BRAF V600E; ERK; Mcl-1; everolimus; mTOR.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures





References
-
- Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433–439. - PubMed
-
- Kim DD, Eng C. The promise of mTOR inhibitors in the treatment of colorectal cancer. Expert opinion on investigational drugs. 2012;21:1775–1788. - PubMed
-
- Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, Gray N, Barletta JA, Guo Y, Swanson SJ, Ruan DT, Hanna GJ, Haddad RI, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. The New England journal of medicine. 2014;371:1426–1433. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
