

Loss of function of the retinoid-related nuclear receptor (RORB) gene and epilepsy
- PMID: 27352968
- PMCID: PMC5117930
- DOI: 10.1038/ejhg.2016.80
Loss of function of the retinoid-related nuclear receptor (RORB) gene and epilepsy
Abstract
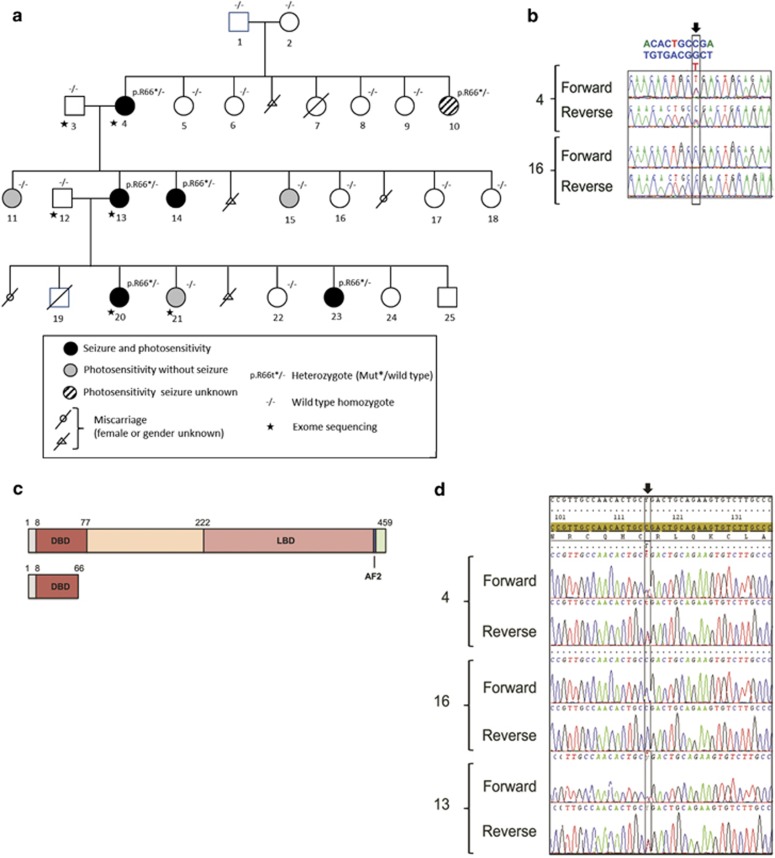
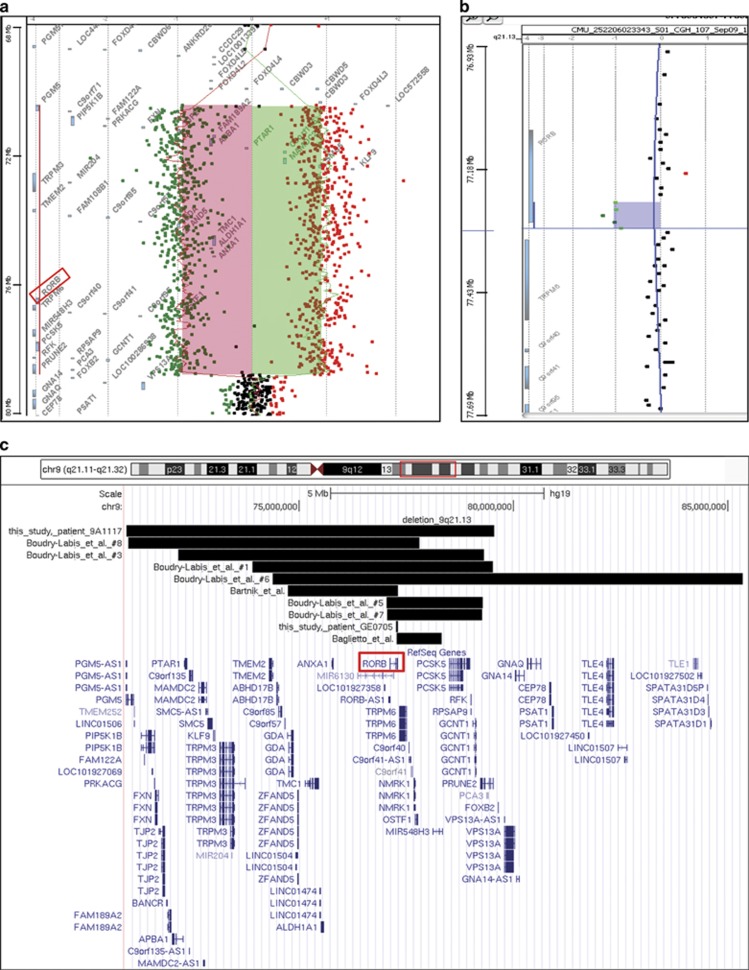
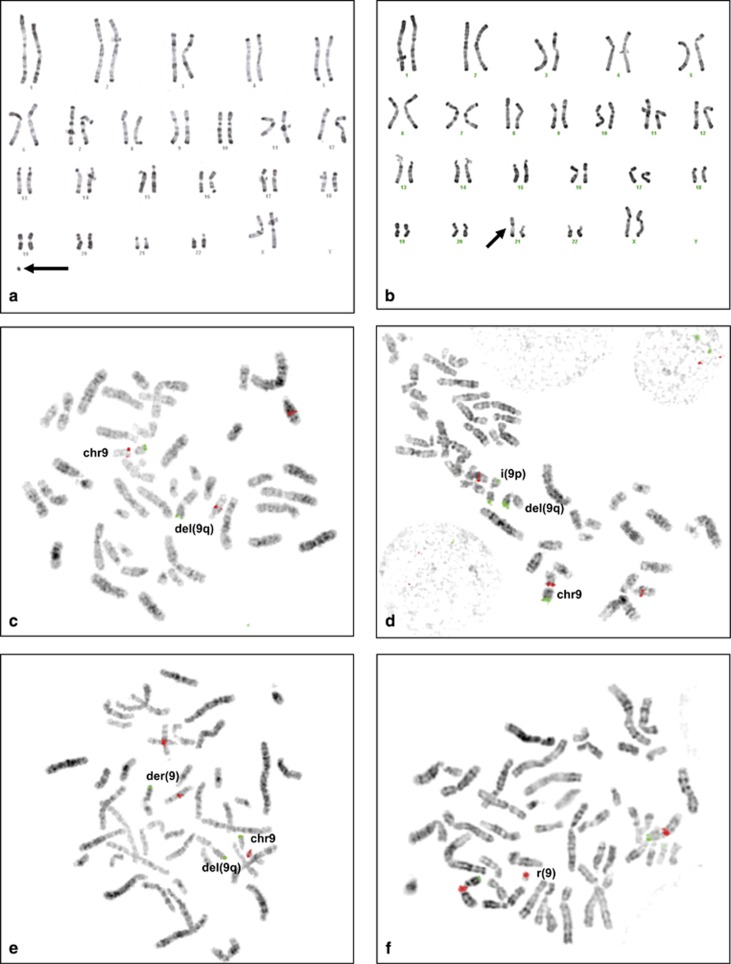
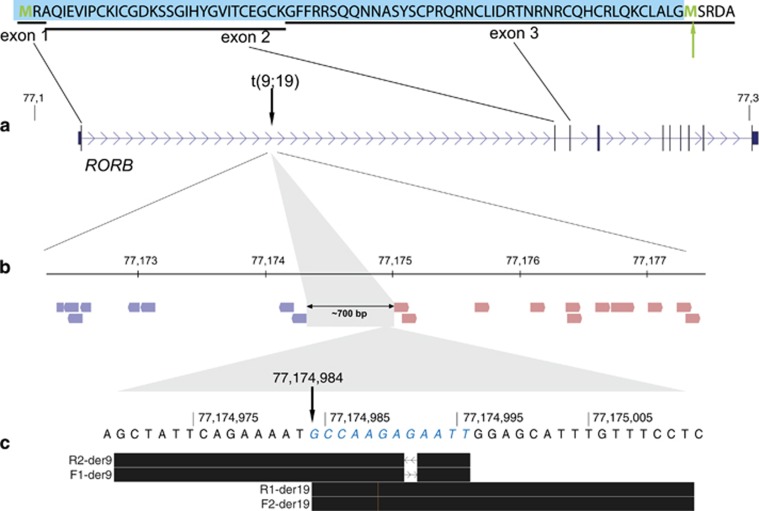
Genetic generalized epilepsy (GGE), formerly known as idiopathic generalized epilepsy, is the most common form of epilepsy and is thought to have predominant genetic etiology. GGE are clinically characterized by absence, myoclonic, or generalized tonic-clonic seizures with electroencephalographic pattern of bilateral, synchronous, and symmetrical spike-and-wave discharges. Despite their strong heritability, the genetic basis of generalized epilepsies remains largely elusive. Nevertheless, recent advances in genetic technology have led to the identification of numerous genes and genomic defects in various types of epilepsies in the past few years. In the present study, we performed whole-exome sequencing in a family with GGE consistent with the diagnosis of eyelid myoclonia with absences. We found a nonsense variant (c.196C>T/p.(Arg66*)) in RORB, which encodes the beta retinoid-related orphan nuclear receptor (RORβ), in four affected family members. In addition, two de novo variants (c.218T>C/p.(Leu73Pro); c.1249_1251delACG/p.(Thr417del)) were identified in sporadic patients by trio-based exome sequencing. We also found two de novo deletions in patients with behavioral and cognitive impairment and epilepsy: a 52-kb microdeletion involving exons 5-10 of RORB and a larger 9q21-microdeletion. Furthermore, we identified a patient with intellectual disability and a balanced translocation where one breakpoint truncates RORB and refined the phenotype of a recently reported patient with RORB deletion. Our data support the role of RORB gene variants/CNVs in neurodevelopmental disorders including epilepsy, and especially in generalized epilepsies with predominant absence seizures.
Conflict of interest statement
Dr N Tommerup receives funding from the University of Copenhagen (UCPH), Lundbeck Foundation, and The Danish Council for Independent Research – Medical Sciences. Dr KL Helbig, Dr S Tang, Dr DN Shinde, Dr R Huether, and Dr HM Lu are employed by and receives a salary from Ambry Genetics. Exome sequencing is among its commercially available tests. Dr S Coulbaut and Dr M Abbas are UCB Pharma employees. Dr Scheffer reports grants from NHMRC and NIH during the conduct of the study: Annals of Neurology, Epileptic Disorders, and Neurology; personal fees from UCB, Athena Diagnostics, Transgenomics, and GlaxoSmithKline, outside the submitted work; in addition, Dr Scheffer has a patent A Diagnostic Method for Epilepsy with royalties paid. Hannah Stamberger is PhD fellow of the Fund for Scientific Research Flanders. Dr Heather Mefford receives funding from the National Institutes of Health. Dr Ingo Helbig is supported by intramural funds of the University of Kiel, by a grant from the German Research Foundation (HE5415/3-1) within the EuroEPINOMICS framework of the European Science Foundation and grants of the German Research Foundation (DFG, HE5415/5-1, HE 5415/6-1), German Ministry for Education and Research (01DH12033, MAR 10/012), and German chapter of the International League against Epilepsy (DGfE). He is also supported by the Children's Hospital of Philadelphia (CHOP) with the Genomics Research Initiative Network (GRIN). Dr Szepetowski reports grants from French National Research Agency (ANR), UCB-Pharma France, and European Union FP7 during the conduct of the study. All the other authors declare no conflict of interest.
Figures
References
-
- Nabbout R, Scheffer IE: Genetics of idiopathic epilepsies. Handb Clin Neurol 2013; 111: 567–578. - PubMed
-
- Dibbens LM, Heron SE, Mulley JC: A polygenic heterogeneity model for common epilepsies with complex genetics. Genes Brain Behav 2007; 6: 593–597. - PubMed
-
- Scheffer IE, Mefford HC: Epilepsy: beyond the single nucleotide variant in epilepsy genetics. Nat Rev Neurol 2014; 10: 490–491. - PubMed
-
- Rubboli G, Gardella E, Capovilla G: Idiopathic generalized epilepsy (IGE) syndromes in development: IGE with absences of early childhood, IGE with phantom absences, and perioral myoclonia with absences. Epilepsia 2009; 50 (Suppl 5): 24–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
