

Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction
- PMID: 27353089
- PMCID: PMC4925026
- DOI: 10.1084/jem.20160081
Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction
Abstract
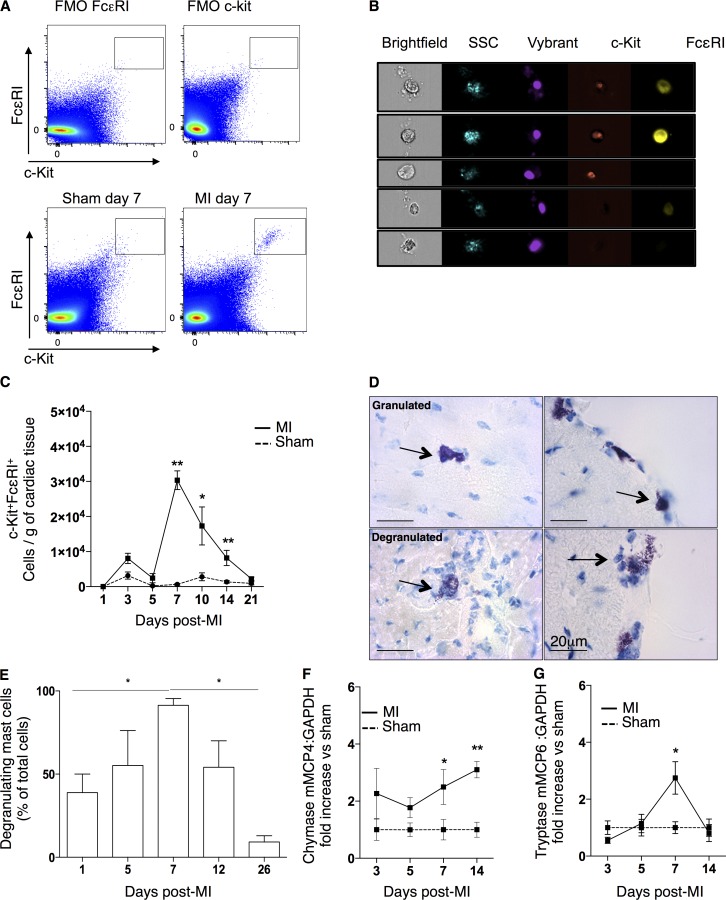
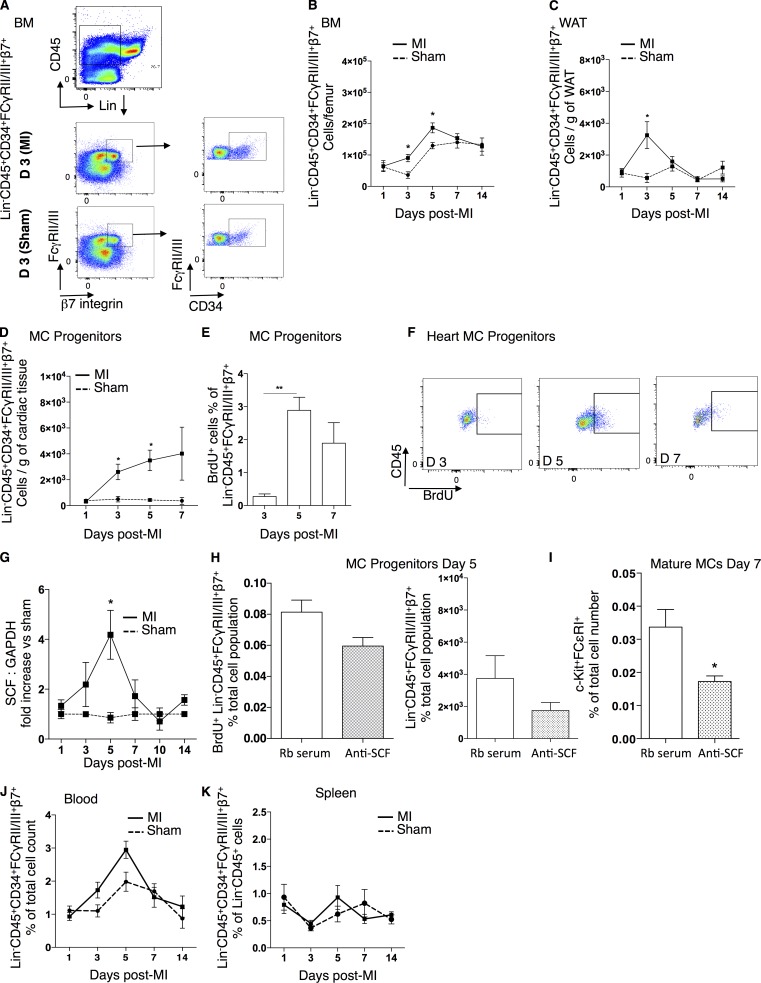
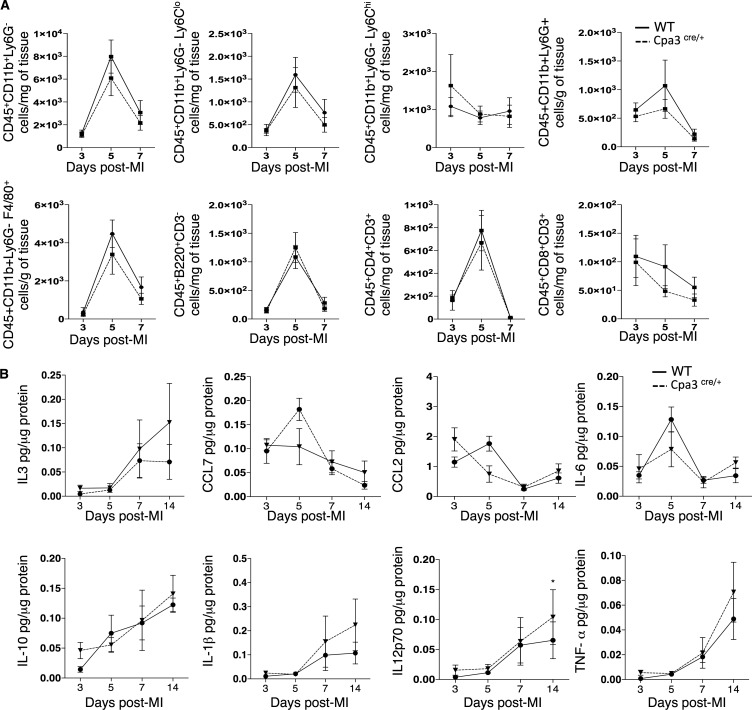
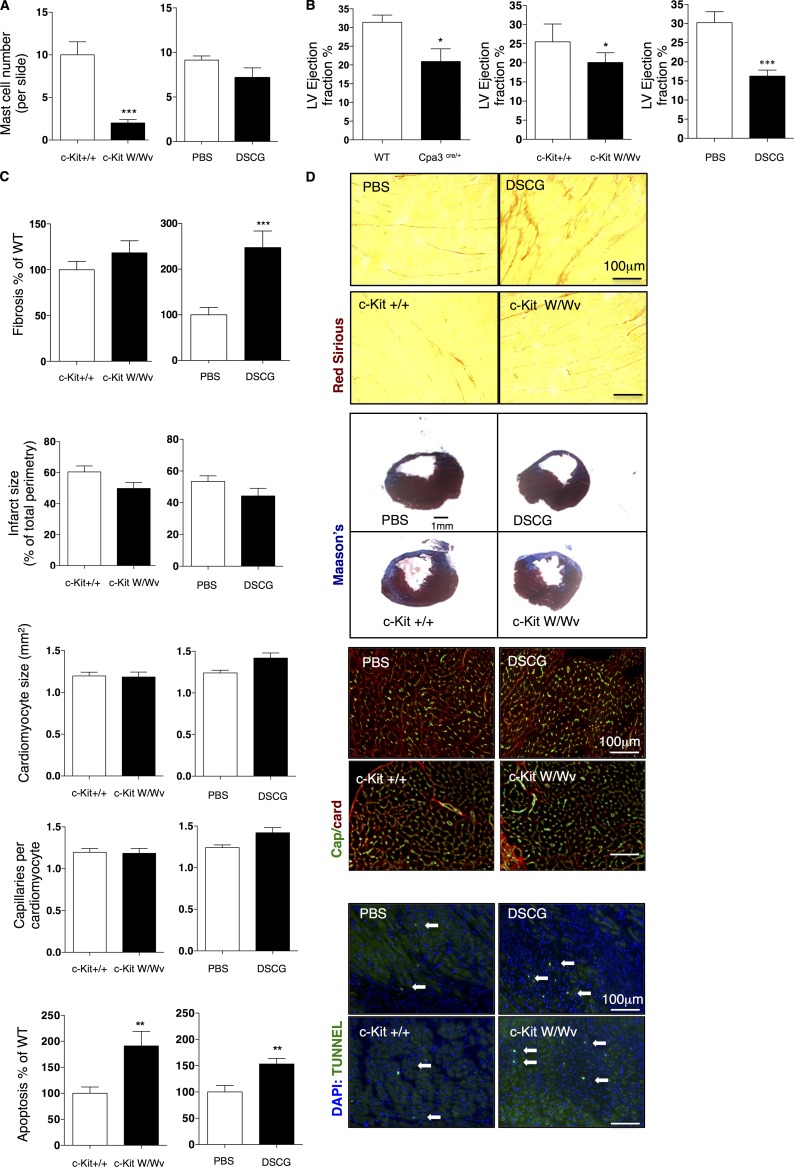
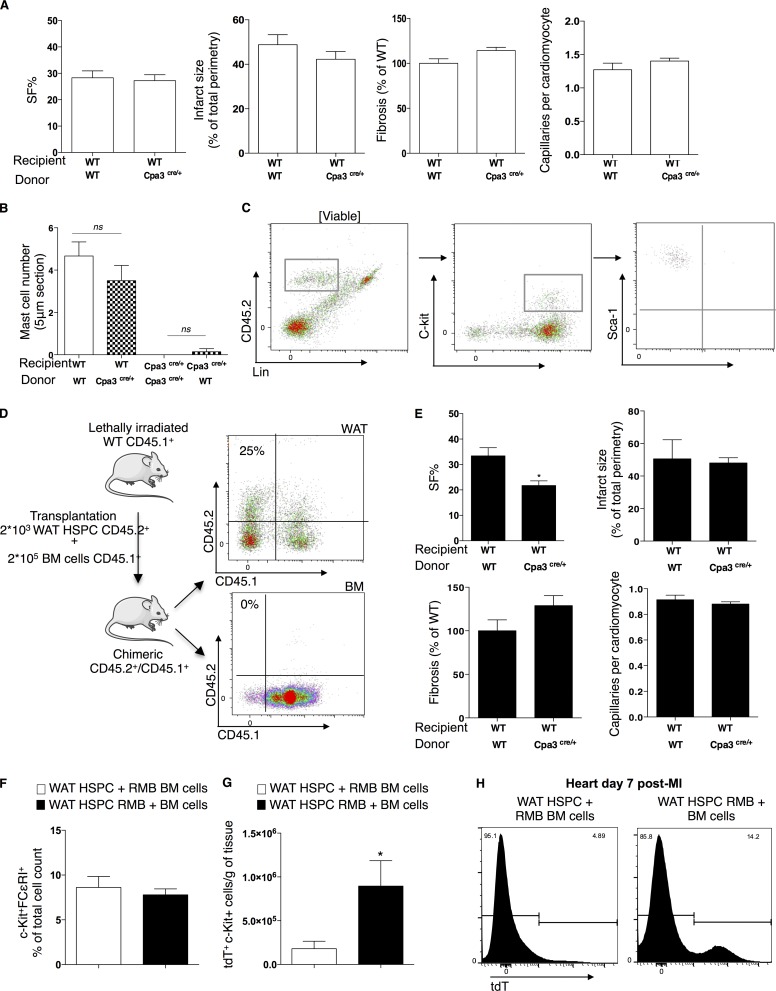
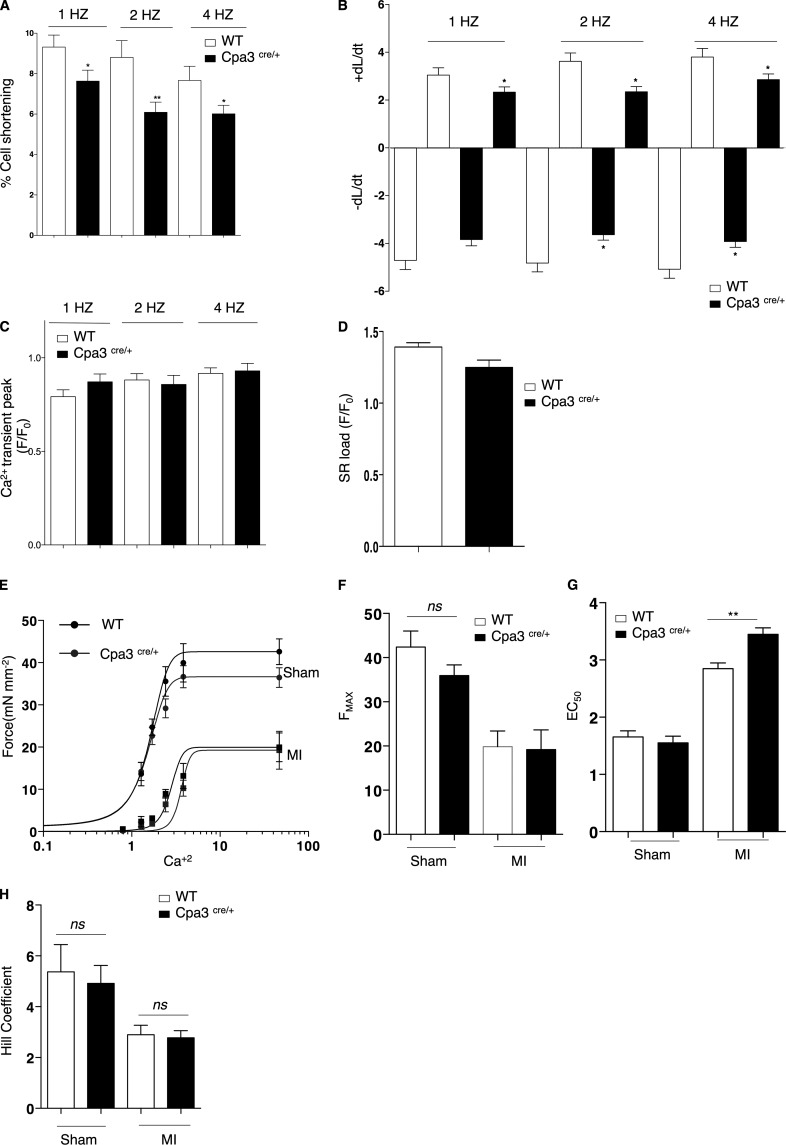
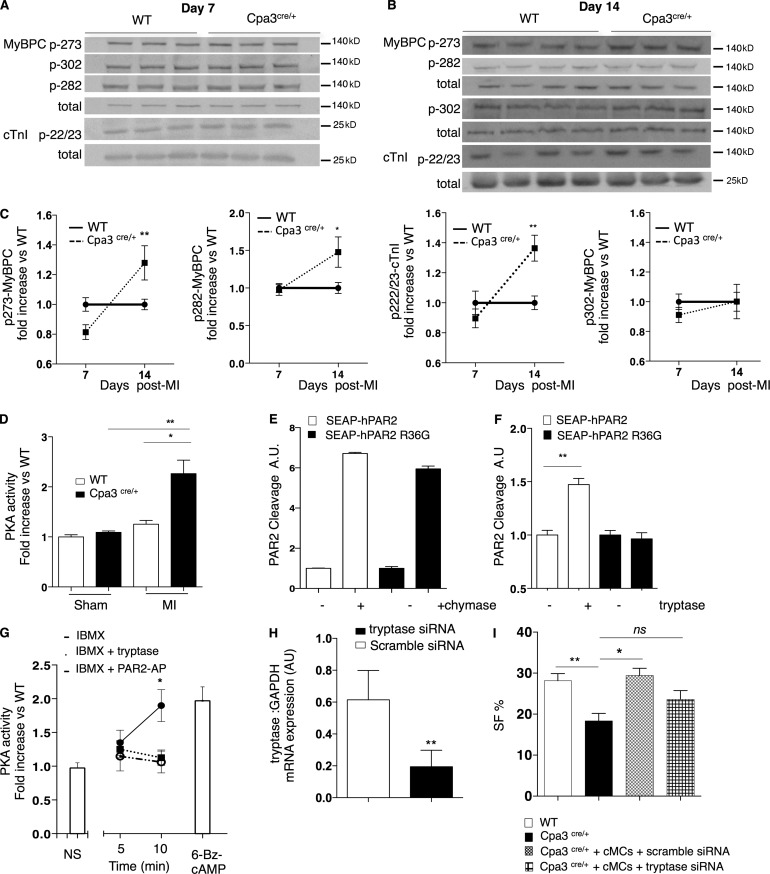
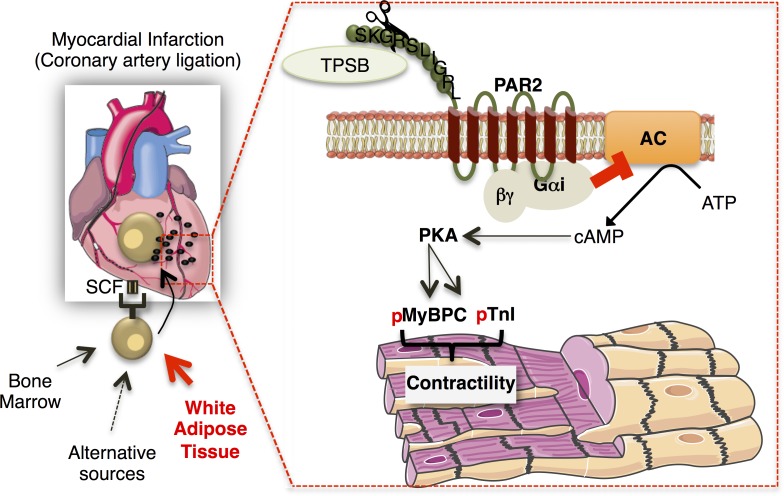
Acute myocardial infarction (MI) is a severe ischemic disease responsible for heart failure and sudden death. Inflammatory cells orchestrate postischemic cardiac remodeling after MI. Studies using mice with defective mast/stem cell growth factor receptor c-Kit have suggested key roles for mast cells (MCs) in postischemic cardiac remodeling. Because c-Kit mutations affect multiple cell types of both immune and nonimmune origin, we addressed the impact of MCs on cardiac function after MI, using the c-Kit-independent MC-deficient (Cpa3(Cre/+)) mice. In response to MI, MC progenitors originated primarily from white adipose tissue, infiltrated the heart, and differentiated into mature MCs. MC deficiency led to reduced postischemic cardiac function and depressed cardiomyocyte contractility caused by myofilament Ca(2+) desensitization. This effect correlated with increased protein kinase A (PKA) activity and hyperphosphorylation of its targets, troponin I and myosin-binding protein C. MC-specific tryptase was identified to regulate PKA activity in cardiomyocytes via protease-activated receptor 2 proteolysis. This work reveals a novel function for cardiac MCs modulating cardiomyocyte contractility via alteration of PKA-regulated force-Ca(2+) interactions in response to MI. Identification of this MC-cardiomyocyte cross-talk provides new insights on the cellular and molecular mechanisms regulating the cardiac contractile machinery and a novel platform for therapeutically addressable regulators.
©2016 Ngkelo et al.
Figures

References
-
- Akers, I.A., Parsons M., Hill M.R., Hollenberg M.D., Sanjar S., Laurent G.J., and McAnulty R.J.. 2000. Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 278:L193–L201. - PubMed
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
