

Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides
- PMID: 27353631
- PMCID: PMC5026416
- DOI: 10.1021/acs.chemrev.6b00180
Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides
Abstract
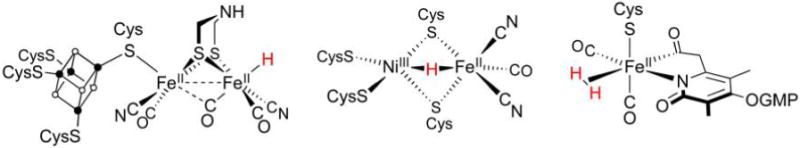
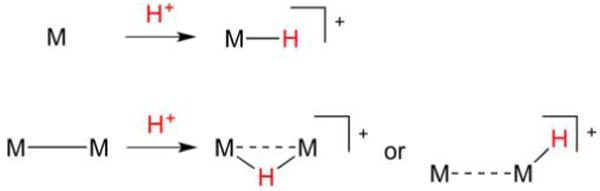
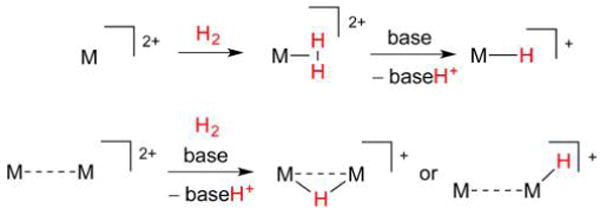
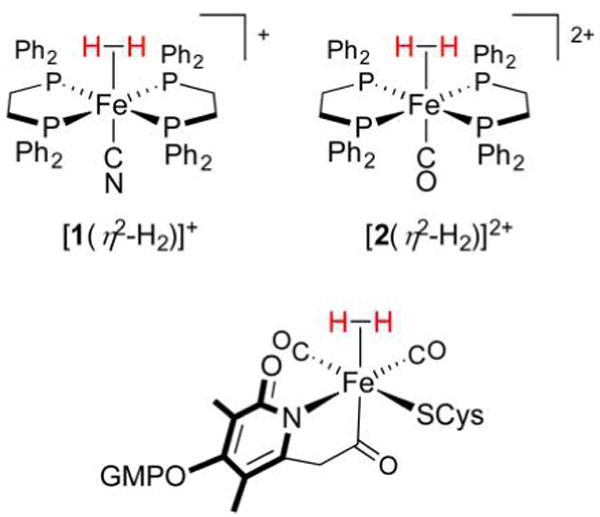
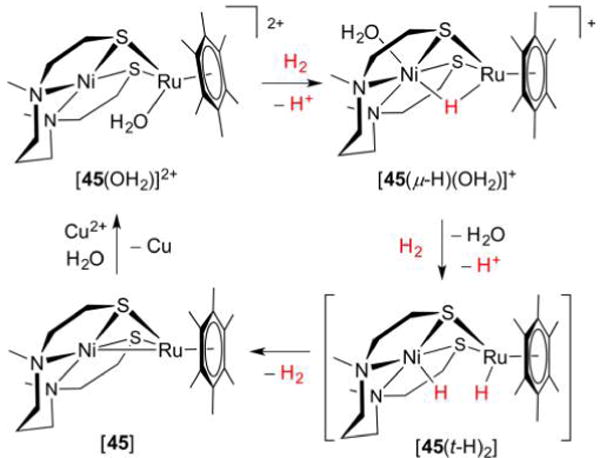
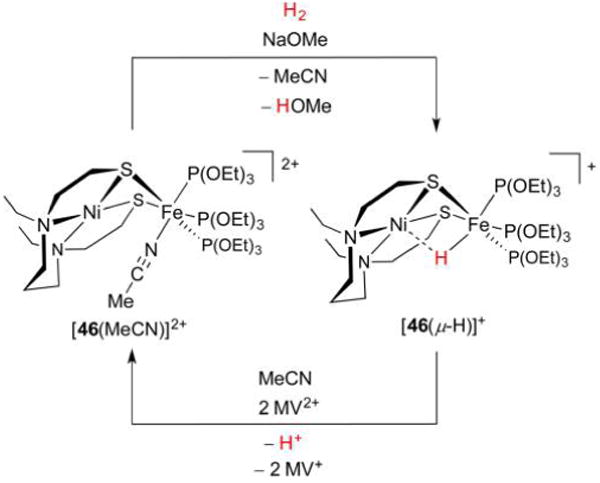
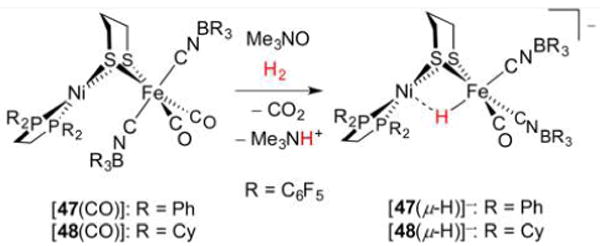
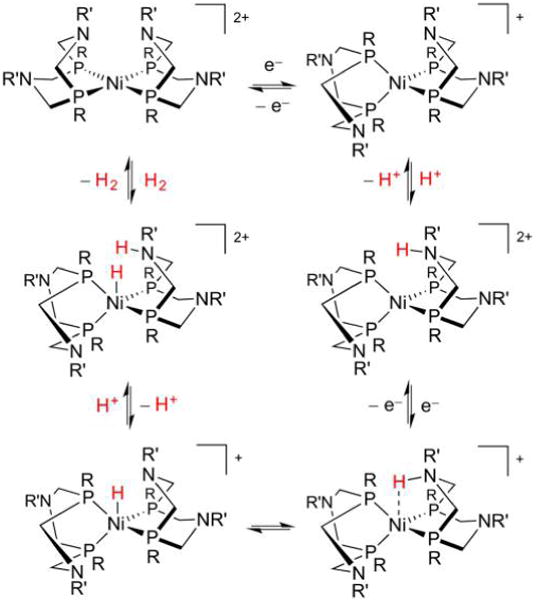
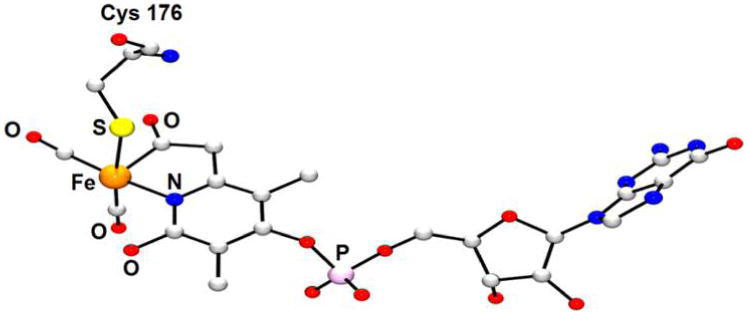
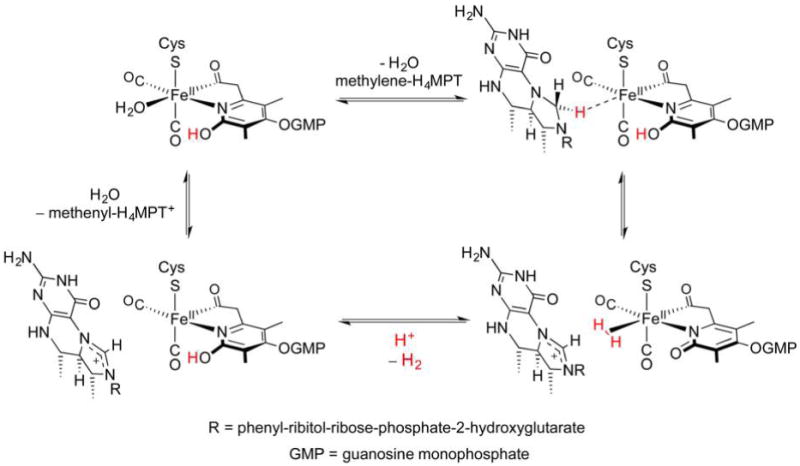
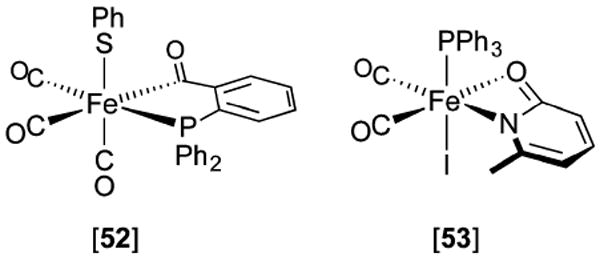
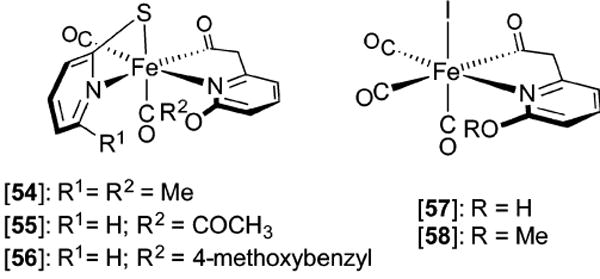
Hydrogenase enzymes efficiently process H2 and protons at organometallic FeFe, NiFe, or Fe active sites. Synthetic modeling of the many H2ase states has provided insight into H2ase structure and mechanism, as well as afforded catalysts for the H2 energy vector. Particularly important are hydride-bearing states, with synthetic hydride analogues now known for each hydrogenase class. These hydrides are typically prepared by protonation of low-valent cores. Examples of FeFe and NiFe hydrides derived from H2 have also been prepared. Such chemistry is more developed than mimicry of the redox-inactive monoFe enzyme, although functional models of the latter are now emerging. Advances in physical and theoretical characterization of H2ase enzymes and synthetic models have proven key to the study of hydrides in particular, and will guide modeling efforts toward more robust and active species optimized for practical applications.
Conflict of interest statement
Notes The authors declare no competing financial interest.
Figures

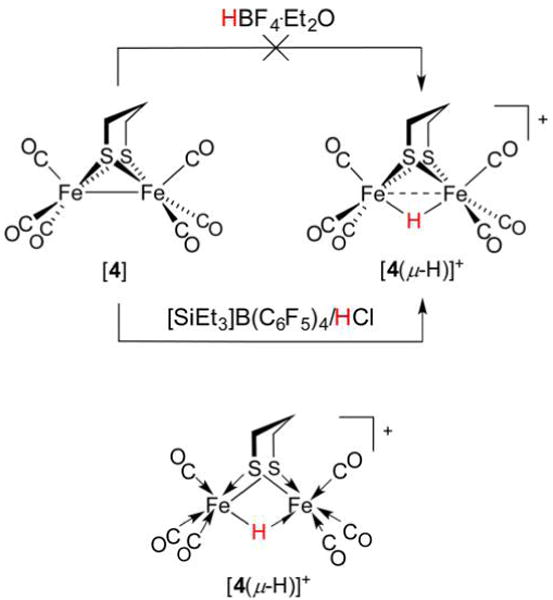

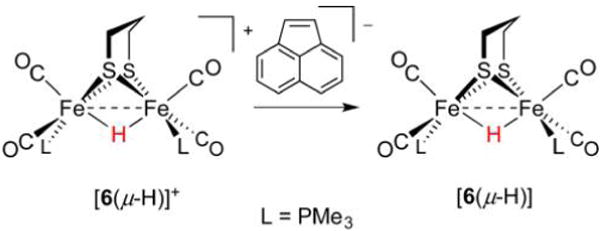
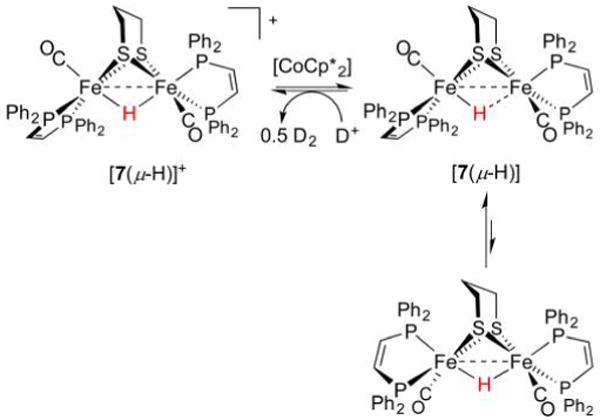
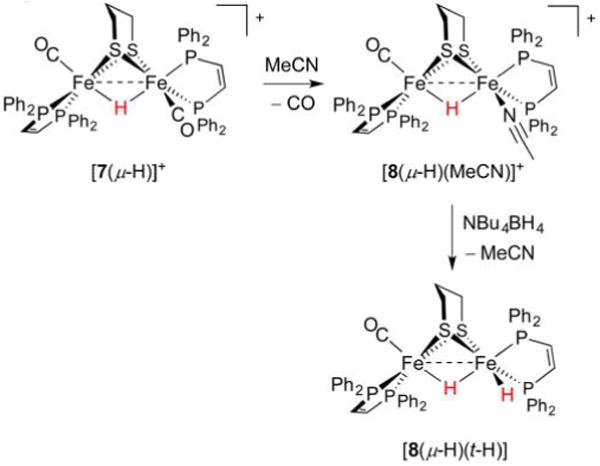
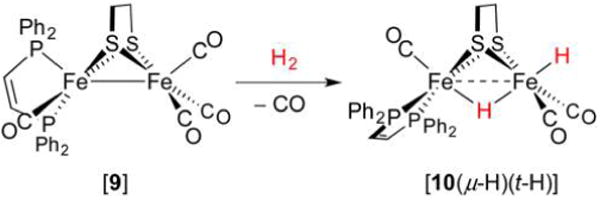
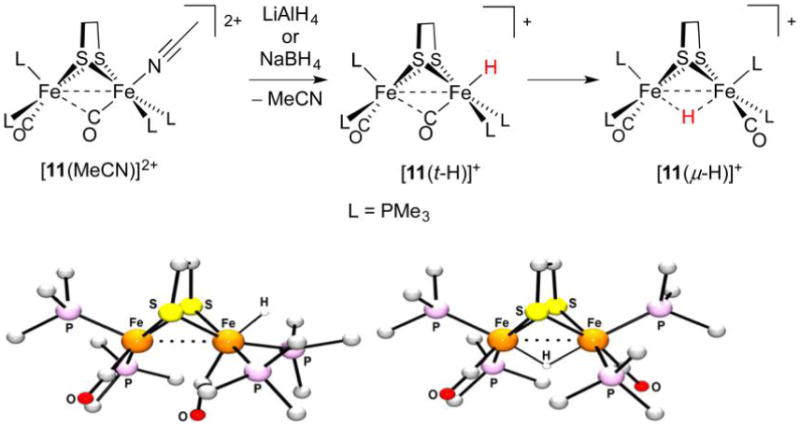
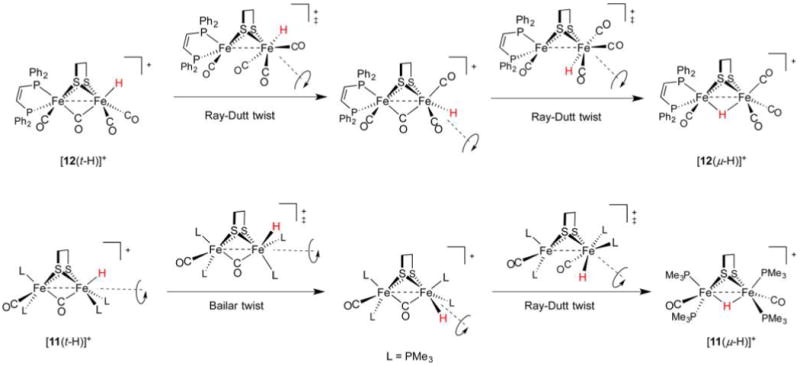
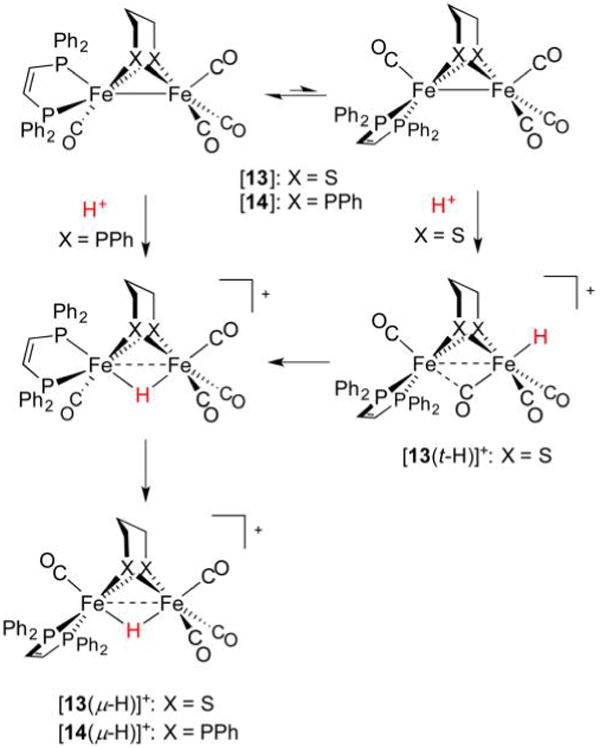
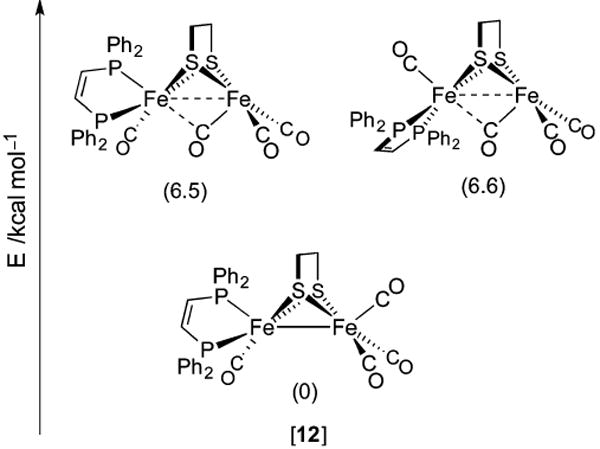
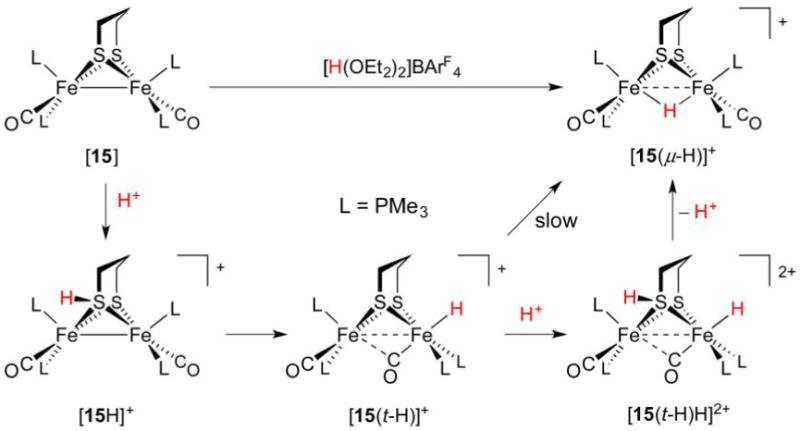

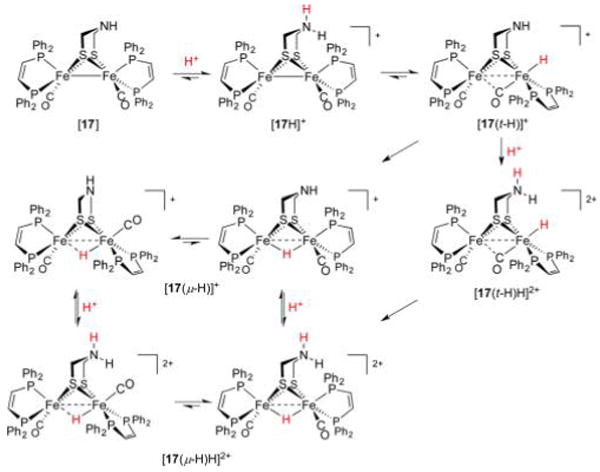
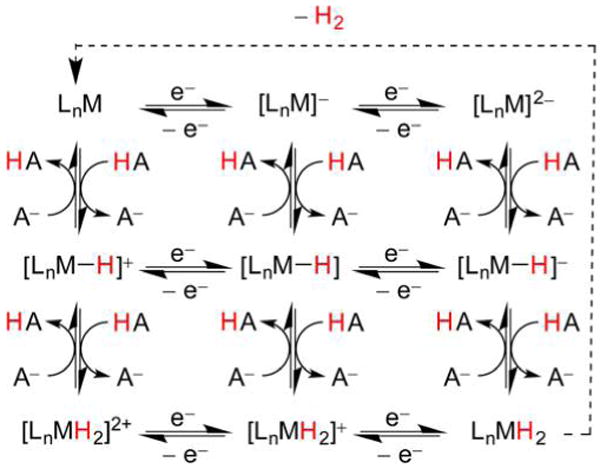
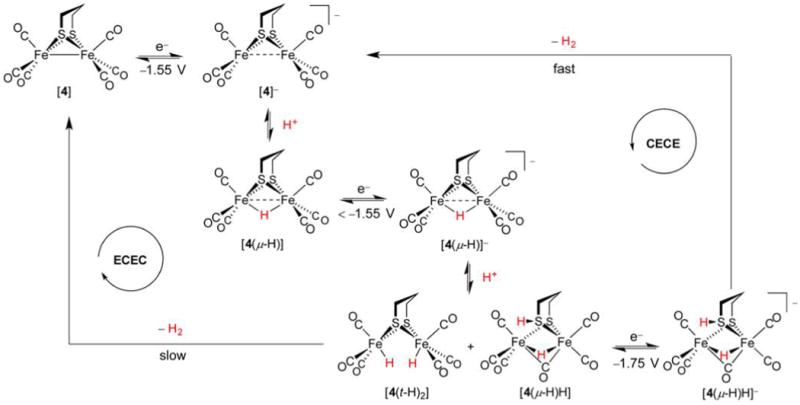



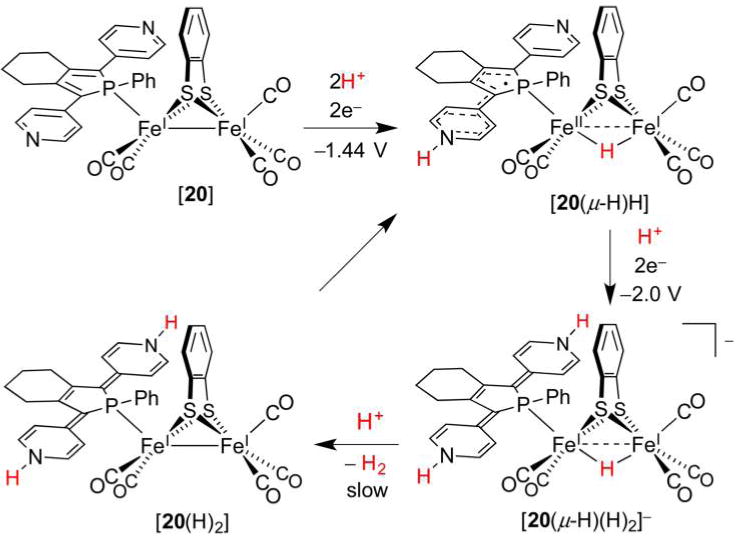
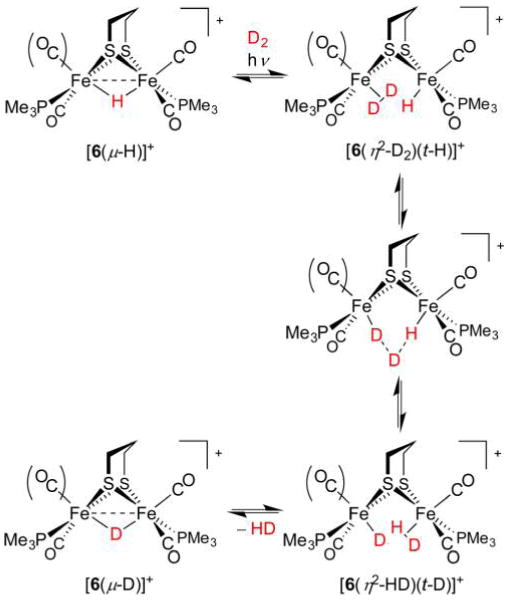
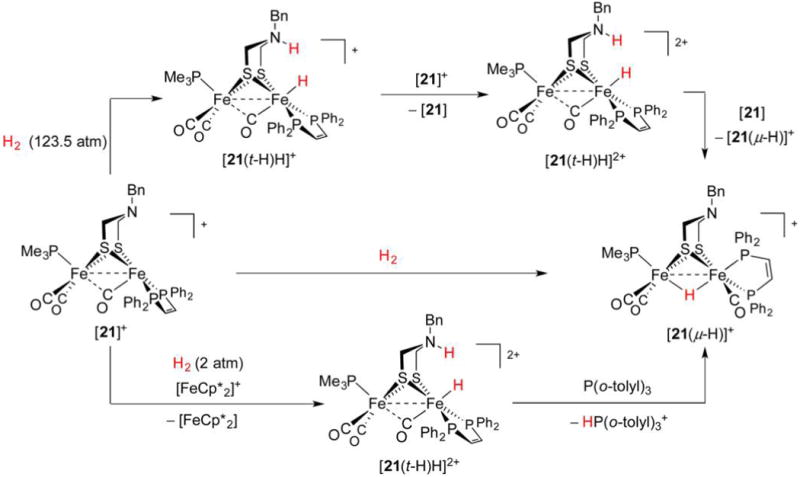
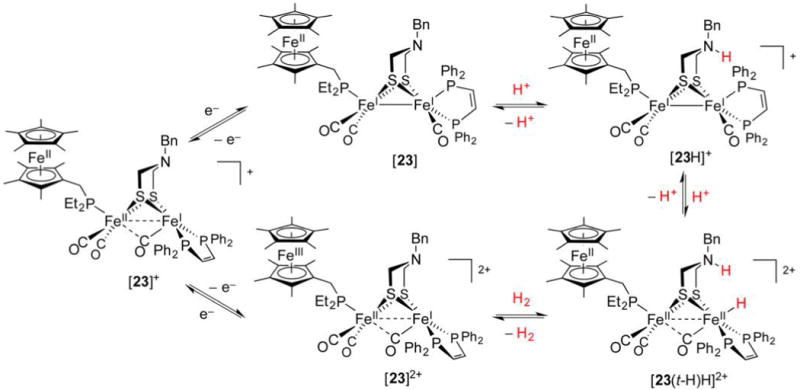

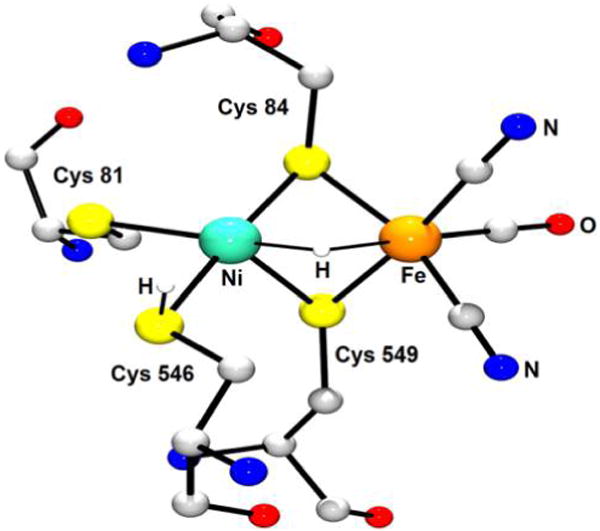
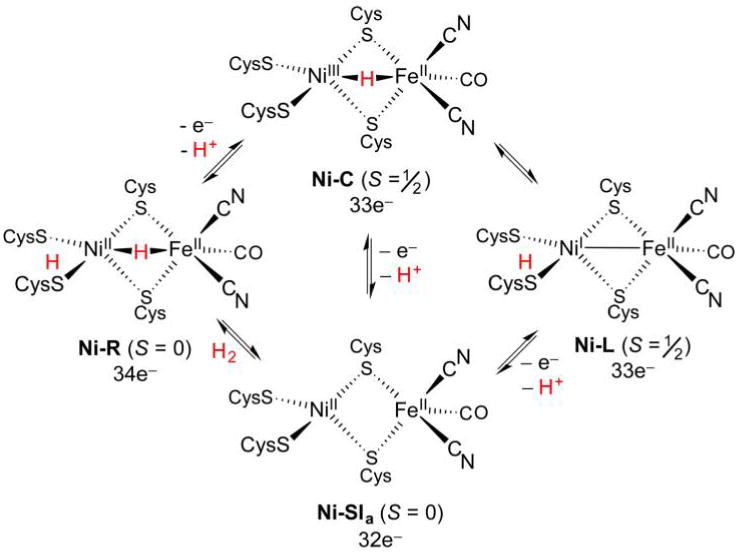
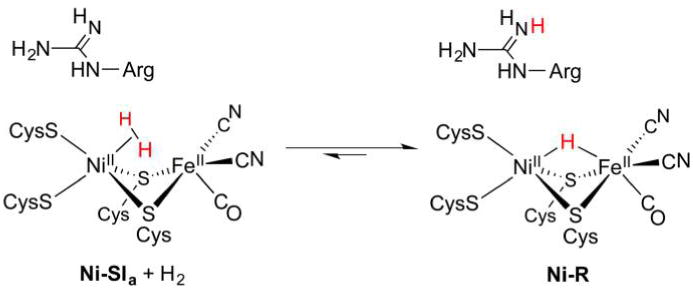

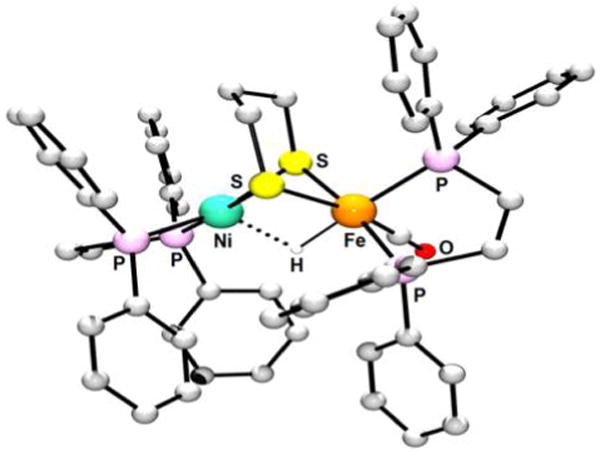
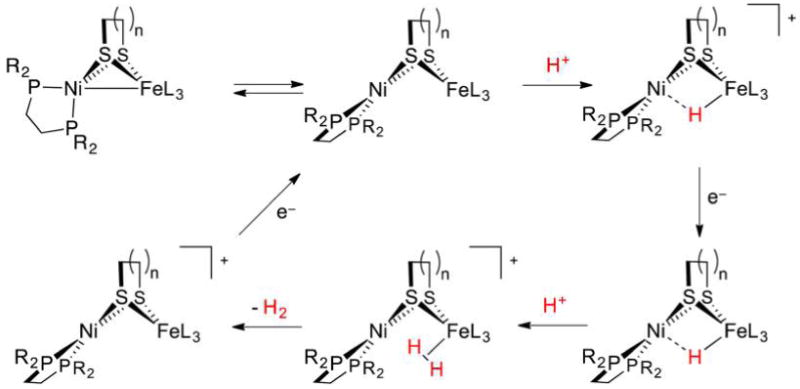
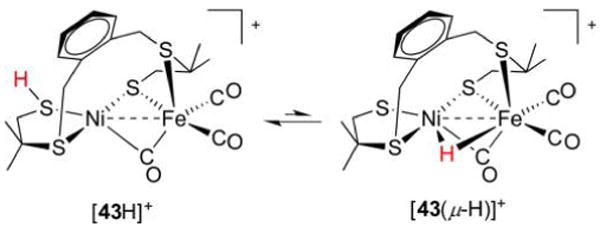
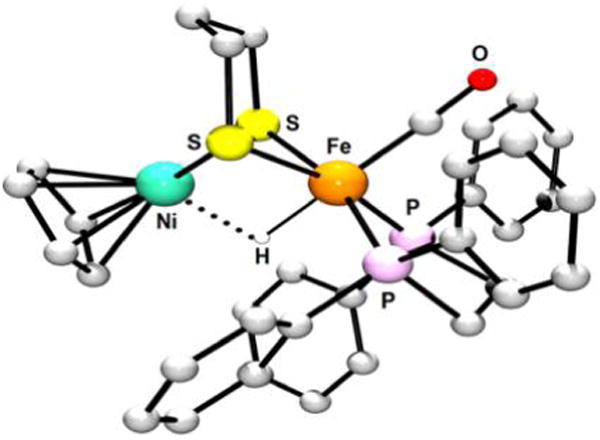


References
-
- Immerdorf H. Contributions to the solution of the “nitrogen question”. Landwirtsch Jahrb. 1892;21:281.
-
- Tausz J, Donath H. On the oxidation of hydrogen and hydrocarbons by bacteria. Hoppe-Seyler’s Z Physiol Chem. 1930;190:141.
-
- Elsden SR. Hydrogenase 1931–1981. Trends Biochem Sci. 1981;6:251–253.
-
- Stripp ST, Happe T. How algae produce hydrogen—news from the photosynthetic hydrogenase. Dalton Trans. 2009:9960–9969. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
