

β-arrestin-biased signaling through the β2-adrenergic receptor promotes cardiomyocyte contraction
- PMID: 27354517
- PMCID: PMC4948363
- DOI: 10.1073/pnas.1606267113
β-arrestin-biased signaling through the β2-adrenergic receptor promotes cardiomyocyte contraction
Abstract
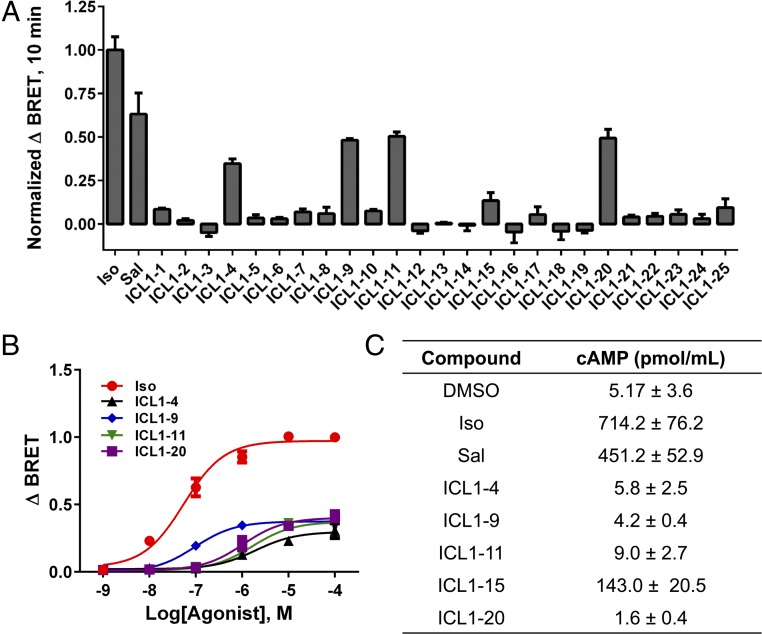
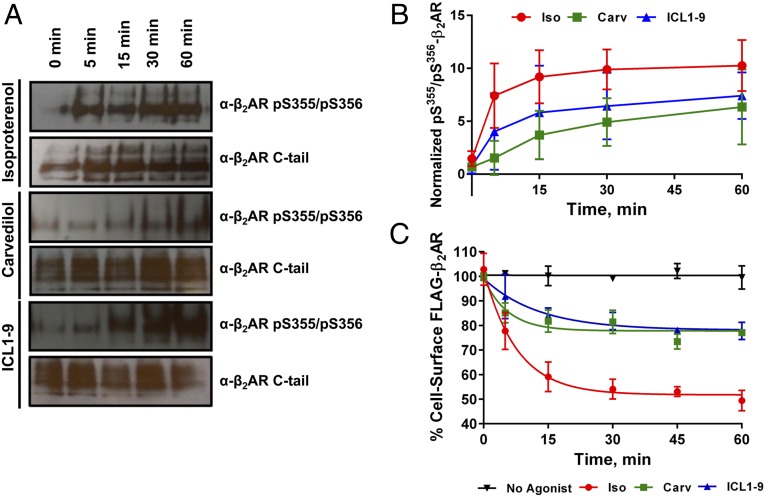
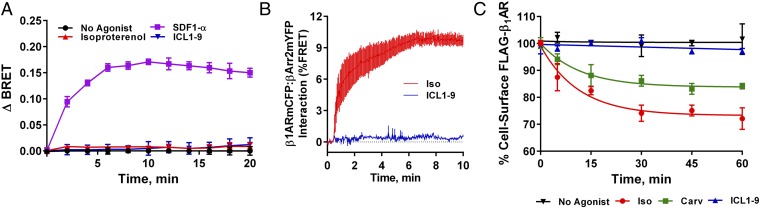
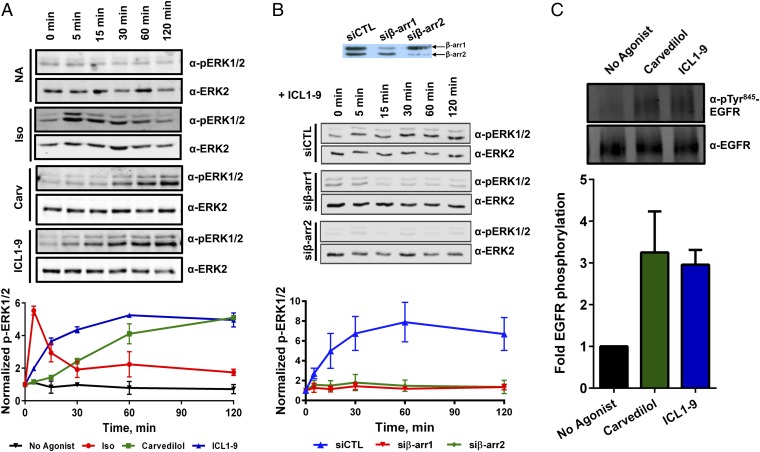
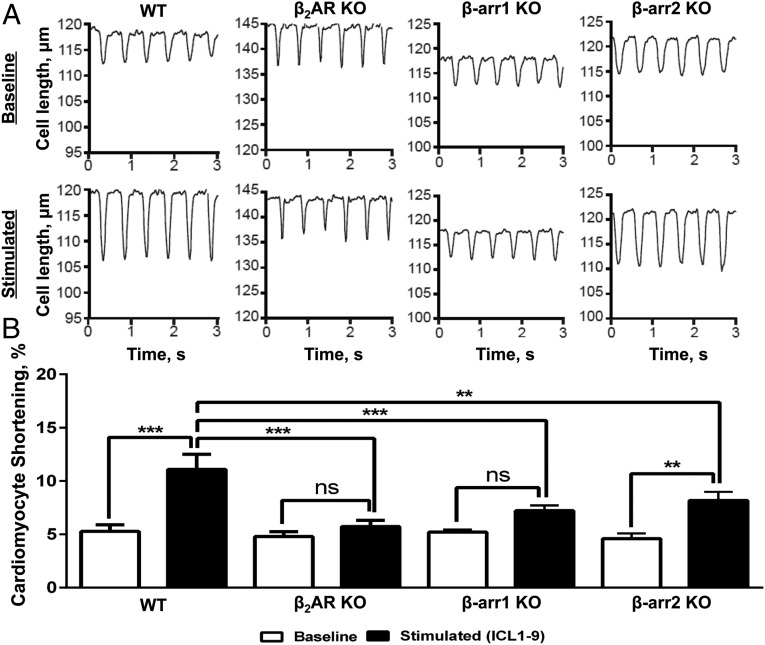
β-adrenergic receptors (βARs) are critical regulators of acute cardiovascular physiology. In response to elevated catecholamine stimulation during development of congestive heart failure (CHF), chronic activation of Gs-dependent β1AR and Gi-dependent β2AR pathways leads to enhanced cardiomyocyte death, reduced β1AR expression, and decreased inotropic reserve. β-blockers act to block excessive catecholamine stimulation of βARs to decrease cellular apoptotic signaling and normalize β1AR expression and inotropy. Whereas these actions reduce cardiac remodeling and mortality outcomes, the effects are not sustained. Converse to G-protein-dependent signaling, β-arrestin-dependent signaling promotes cardiomyocyte survival. Given that β2AR expression is unaltered in CHF, a β-arrestin-biased agonist that operates through the β2AR represents a potentially useful therapeutic approach. Carvedilol, a currently prescribed nonselective β-blocker, has been classified as a β-arrestin-biased agonist that can inhibit basal signaling from βARs and also stimulate cell survival signaling pathways. To understand the relative contribution of β-arrestin bias to the efficacy of select β-blockers, a specific β-arrestin-biased pepducin for the β2AR, intracellular loop (ICL)1-9, was used to decouple β-arrestin-biased signaling from occupation of the orthosteric ligand-binding pocket. With similar efficacy to carvedilol, ICL1-9 was able to promote β2AR phosphorylation, β-arrestin recruitment, β2AR internalization, and β-arrestin-biased signaling. Interestingly, ICL1-9 was also able to induce β2AR- and β-arrestin-dependent and Ca(2+)-independent contractility in primary adult murine cardiomyocytes, whereas carvedilol had no efficacy. Thus, ICL1-9 is an effective tool to access a pharmacological profile stimulating cardioprotective signaling and inotropic effects through the β2AR and serves as a model for the next generation of cardiovascular drug development.
Keywords: GPCR; arrestin; carvedilol; heart failure; pepducin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
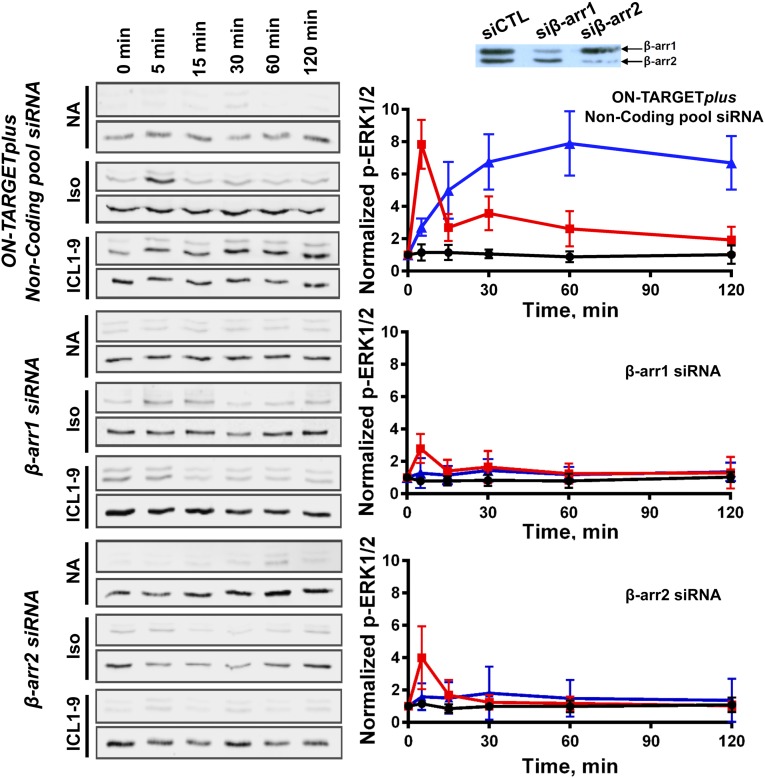
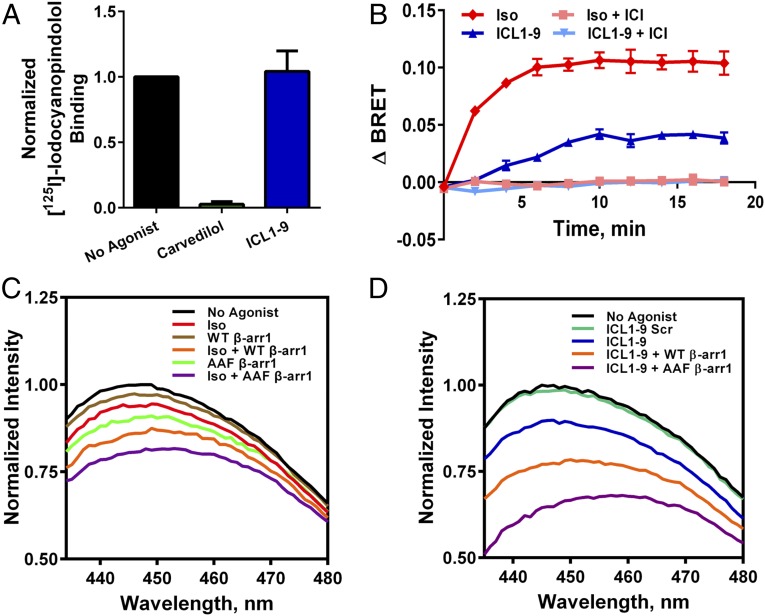
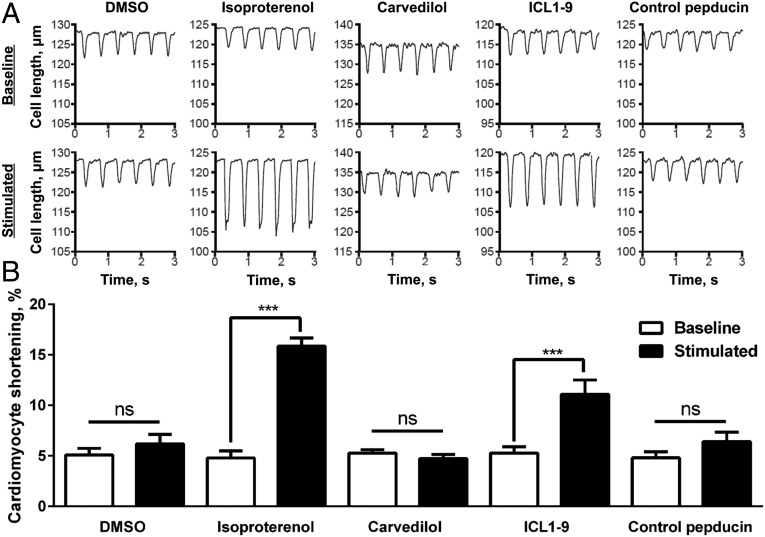
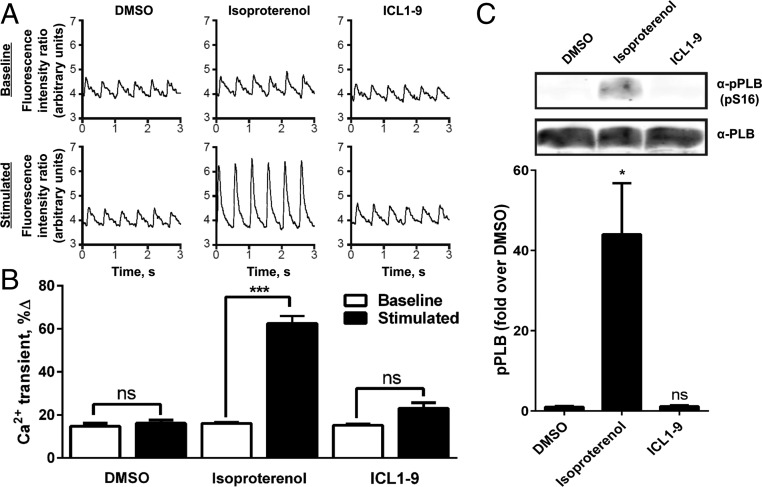

References
-
- Javed U, Deedwania PC. Beta-adrenergic blockers for chronic heart failure. Cardiol Rev. 2009;17(6):287–292. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
